Coverage Report
File Sequence.java
Coverage histogram
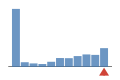
0% of files have more coverage
Classes
Class |
Line # |
Actions |
|||
|---|---|---|---|---|---|
| Sequence | 50 | 660 | 348 | ||
| Sequence.DBModList | 62 | 1 | 1 |
Contributing tests
This file is covered by 760 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | package jalview.datamodel; | |
| 22 | ||
| 23 | import java.util.ArrayList; | |
| 24 | import java.util.Arrays; | |
| 25 | import java.util.BitSet; | |
| 26 | import java.util.Collection; | |
| 27 | import java.util.Collections; | |
| 28 | import java.util.Enumeration; | |
| 29 | import java.util.Iterator; | |
| 30 | import java.util.List; | |
| 31 | import java.util.ListIterator; | |
| 32 | import java.util.Vector; | |
| 33 | ||
| 34 | import fr.orsay.lri.varna.models.rna.RNA; | |
| 35 | import jalview.analysis.AlignSeq; | |
| 36 | import jalview.analysis.AlignmentUtils; | |
| 37 | import jalview.analysis.SeqsetUtils; | |
| 38 | import jalview.datamodel.features.SequenceFeatures; | |
| 39 | import jalview.datamodel.features.SequenceFeaturesI; | |
| 40 | import jalview.util.Comparison; | |
| 41 | import jalview.util.DBRefUtils; | |
| 42 | import jalview.util.MapList; | |
| 43 | import jalview.util.StringUtils; | |
| 44 | import jalview.ws.datamodel.alphafold.MappableContactMatrix; | |
| 45 | ||
| 46 | /** | |
| 47 | * | |
| 48 | * Implements the SequenceI interface for a char[] based sequence object | |
| 49 | */ | |
| 50 | public class Sequence extends ASequence implements SequenceI | |
| 51 | { | |
| 52 | ||
| 53 | /** | |
| 54 | * A subclass that gives us access to modCount, which tracks whether there | |
| 55 | * have been any changes. We use this to update | |
| 56 | * | |
| 57 | * @author hansonr | |
| 58 | * | |
| 59 | * @param <T> | |
| 60 | */ | |
| 61 | @SuppressWarnings("serial") | |
| 62 | public class DBModList<T> extends ArrayList<DBRefEntry> | |
| 63 | { | |
| 64 | ||
| 65 | 581 |  protected int getModCount() protected int getModCount() |
| 66 | { | |
| 67 | 581 | return modCount; |
| 68 | } | |
| 69 | ||
| 70 | } | |
| 71 | ||
| 72 | SequenceI datasetSequence; | |
| 73 | ||
| 74 | private String name; | |
| 75 | ||
| 76 | private char[] sequence; | |
| 77 | ||
| 78 | private String description; | |
| 79 | ||
| 80 | private int start; | |
| 81 | ||
| 82 | private int end; | |
| 83 | ||
| 84 | private Vector<PDBEntry> pdbIds; | |
| 85 | ||
| 86 | private String vamsasId; | |
| 87 | ||
| 88 | private DBModList<DBRefEntry> dbrefs; // controlled access | |
| 89 | ||
| 90 | /** | |
| 91 | * a flag to let us know that elements have changed in dbrefs | |
| 92 | * | |
| 93 | * @author Bob Hanson | |
| 94 | */ | |
| 95 | private int refModCount = 0; | |
| 96 | ||
| 97 | private RNA rna; | |
| 98 | ||
| 99 | /** | |
| 100 | * This annotation is displayed below the alignment but the positions are tied | |
| 101 | * to the residues of this sequence | |
| 102 | * | |
| 103 | * TODO: change to List<> | |
| 104 | */ | |
| 105 | private Vector<AlignmentAnnotation> annotation; | |
| 106 | ||
| 107 | private SequenceFeaturesI sequenceFeatureStore; | |
| 108 | ||
| 109 | /* | |
| 110 | * A cursor holding the approximate current view position to the sequence, | |
| 111 | * as determined by findIndex or findPosition or findPositions. | |
| 112 | * Using a cursor as a hint allows these methods to be more performant for | |
| 113 | * large sequences. | |
| 114 | */ | |
| 115 | private SequenceCursor cursor; | |
| 116 | ||
| 117 | /* | |
| 118 | * A number that should be incremented whenever the sequence is edited. | |
| 119 | * If the value matches the cursor token, then we can trust the cursor, | |
| 120 | * if not then it should be recomputed. | |
| 121 | */ | |
| 122 | private int changeCount; | |
| 123 | ||
| 124 | /** | |
| 125 | * Creates a new Sequence object. | |
| 126 | * | |
| 127 | * @param name | |
| 128 | * display name string | |
| 129 | * @param sequence | |
| 130 | * string to form a possibly gapped sequence out of | |
| 131 | * @param start | |
| 132 | * first position of non-gap residue in the sequence | |
| 133 | * @param end | |
| 134 | * last position of ungapped residues (nearly always only used for | |
| 135 | * display purposes) | |
| 136 | */ | |
| 137 | 32910 | public Sequence(String name, String sequence, int start, int end) |
| 138 | { | |
| 139 | 32910 | this(); |
| 140 | 32910 | initSeqAndName(name, sequence.toCharArray(), start, end); |
| 141 | } | |
| 142 | ||
| 143 | 75 | public Sequence(String name, char[] sequence, int start, int end) |
| 144 | { | |
| 145 | 75 | this(); |
| 146 | 75 | initSeqAndName(name, sequence, start, end); |
| 147 | } | |
| 148 | ||
| 149 | /** | |
| 150 | * Stage 1 constructor - assign name, sequence, and set start and end fields. | |
| 151 | * start and end are updated values from name2 if it ends with /start-end | |
| 152 | * | |
| 153 | * @param name2 | |
| 154 | * @param sequence2 | |
| 155 | * @param start2 | |
| 156 | * @param end2 | |
| 157 | */ | |
| 158 | 33539 | protected void initSeqAndName(String name2, char[] sequence2, int start2, |
| 159 | int end2) | |
| 160 | { | |
| 161 | 33539 | this.name = name2; |
| 162 | 33539 | this.sequence = sequence2; |
| 163 | 33539 | this.start = start2; |
| 164 | 33539 | this.end = end2; |
| 165 | 33539 | parseId(); |
| 166 | 33539 | checkValidRange(); |
| 167 | } | |
| 168 | ||
| 169 | /** | |
| 170 | * If 'name' ends in /i-j, where i >= j > 0 are integers, extracts i and j as | |
| 171 | * start and end respectively and removes the suffix from the name | |
| 172 | */ | |
| 173 | 34595 | void parseId() |
| 174 | { | |
| 175 | 34595 | if (name == null) |
| 176 | { | |
| 177 | 1 | jalview.bin.Console.errPrintln( |
| 178 | "POSSIBLE IMPLEMENTATION ERROR: null sequence name passed to constructor."); | |
| 179 | 1 | name = ""; |
| 180 | } | |
| 181 | 34595 | int slashPos = name.lastIndexOf('/'); |
| 182 | 34595 | if (slashPos > -1 && slashPos < name.length() - 1) |
| 183 | { | |
| 184 | 402 | String suffix = name.substring(slashPos + 1); |
| 185 | 402 | String[] range = suffix.split("-"); |
| 186 | 402 | if (range.length == 2) |
| 187 | { | |
| 188 | 399 | try |
| 189 | { | |
| 190 | 399 | int from = Integer.valueOf(range[0]); |
| 191 | 390 | int to = Integer.valueOf(range[1]); |
| 192 | 390 | if (from > 0 && to >= from) |
| 193 | { | |
| 194 | 388 | name = name.substring(0, slashPos); |
| 195 | 388 | setStart(from); |
| 196 | 388 | setEnd(to); |
| 197 | 388 | checkValidRange(); |
| 198 | } | |
| 199 | } catch (NumberFormatException e) | |
| 200 | { | |
| 201 | // leave name unchanged if suffix is invalid | |
| 202 | } | |
| 203 | } | |
| 204 | } | |
| 205 | } | |
| 206 | ||
| 207 | /** | |
| 208 | * Ensures that 'end' is not before the end of the sequence, that is, | |
| 209 | * (end-start+1) is at least as long as the count of ungapped positions. Note | |
| 210 | * that end is permitted to be beyond the end of the sequence data. | |
| 211 | */ | |
| 212 | 44481 | void checkValidRange() |
| 213 | { | |
| 214 | // Note: JAL-774 : | |
| 215 | // http://issues.jalview.org/browse/JAL-774?focusedCommentId=11239&page=com.atlassian.jira.plugin.system.issuetabpanels:comment-tabpanel#comment-11239 | |
| 216 | { | |
| 217 | 44481 | int endRes = 0; |
| 218 | 15012854 | for (int j = 0; j < sequence.length; j++) |
| 219 | { | |
| 220 | 14968382 | if (!Comparison.isGap(sequence[j])) |
| 221 | { | |
| 222 | 6017725 | endRes++; |
| 223 | } | |
| 224 | } | |
| 225 | 44481 | if (endRes > 0) |
| 226 | { | |
| 227 | 40650 | endRes += start - 1; |
| 228 | } | |
| 229 | ||
| 230 | 44481 | if (end < endRes) |
| 231 | { | |
| 232 | 29615 | end = endRes; |
| 233 | } | |
| 234 | } | |
| 235 | ||
| 236 | } | |
| 237 | ||
| 238 | /** | |
| 239 | * default constructor | |
| 240 | */ | |
| 241 | 33264 | private Sequence() |
| 242 | { | |
| 243 | 33264 | sequenceFeatureStore = new SequenceFeatures(); |
| 244 | } | |
| 245 | ||
| 246 | /** | |
| 247 | * Creates a new Sequence object. | |
| 248 | * | |
| 249 | * @param name | |
| 250 | * DOCUMENT ME! | |
| 251 | * @param sequence | |
| 252 | * DOCUMENT ME! | |
| 253 | */ | |
| 254 | 24071 | public Sequence(String name, String sequence) |
| 255 | { | |
| 256 | 24071 | this(name, sequence, 1, -1); |
| 257 | } | |
| 258 | ||
| 259 | /** | |
| 260 | * Creates a new Sequence object with new AlignmentAnnotations but inherits | |
| 261 | * any existing dataset sequence reference. If non exists, everything is | |
| 262 | * copied. | |
| 263 | * | |
| 264 | * @param seq | |
| 265 | * if seq is a dataset sequence, behaves like a plain old copy | |
| 266 | * constructor | |
| 267 | */ | |
| 268 | 275 | public Sequence(SequenceI seq) |
| 269 | { | |
| 270 | 275 | this(seq, seq.getAnnotation()); |
| 271 | } | |
| 272 | ||
| 273 | /** | |
| 274 | * Create a new sequence object with new features, DBRefEntries, and PDBIds | |
| 275 | * but inherits any existing dataset sequence reference, and duplicate of any | |
| 276 | * annotation that is present in the given annotation array. | |
| 277 | * | |
| 278 | * @param seq | |
| 279 | * the sequence to be copied | |
| 280 | * @param alAnnotation | |
| 281 | * an array of annotation including some associated with seq | |
| 282 | */ | |
| 283 | 279 | public Sequence(SequenceI seq, AlignmentAnnotation[] alAnnotation) |
| 284 | { | |
| 285 | 279 | this(); |
| 286 | 279 | initSeqFrom(seq, alAnnotation); |
| 287 | } | |
| 288 | ||
| 289 | /** | |
| 290 | * does the heavy lifting when cloning a dataset sequence, or coping data from | |
| 291 | * dataset to a new derived sequence. | |
| 292 | * | |
| 293 | * @param seq | |
| 294 | * - source of attributes. | |
| 295 | * @param alAnnotation | |
| 296 | * - alignment annotation present on seq that should be copied onto | |
| 297 | * this sequence | |
| 298 | */ | |
| 299 | 554 | protected void initSeqFrom(SequenceI seq, |
| 300 | AlignmentAnnotation[] alAnnotation) | |
| 301 | { | |
| 302 | 554 | char[] oseq = seq.getSequence(); // returns a copy of the array |
| 303 | 554 | initSeqAndName(seq.getName(), oseq, seq.getStart(), seq.getEnd()); |
| 304 | ||
| 305 | 554 | description = seq.getDescription(); |
| 306 | 554 | if (seq != datasetSequence) |
| 307 | { | |
| 308 | 288 | setDatasetSequence(seq.getDatasetSequence()); |
| 309 | } | |
| 310 | ||
| 311 | /* | |
| 312 | * only copy DBRefs and seqfeatures if we really are a dataset sequence | |
| 313 | */ | |
| 314 | 554 | if (datasetSequence == null) |
| 315 | { | |
| 316 | 85 | List<DBRefEntry> dbr = seq.getDBRefs(); |
| 317 | 85 | if (dbr != null) |
| 318 | { | |
| 319 | 4 | for (int i = 0, n = dbr.size(); i < n; i++) |
| 320 | { | |
| 321 | 2 | addDBRef(new DBRefEntry(dbr.get(i))); |
| 322 | } | |
| 323 | } | |
| 324 | ||
| 325 | /* | |
| 326 | * make copies of any sequence features | |
| 327 | */ | |
| 328 | 85 | for (SequenceFeature sf : seq.getSequenceFeatures()) |
| 329 | { | |
| 330 | 195 | addSequenceFeature(new SequenceFeature(sf)); |
| 331 | } | |
| 332 | } | |
| 333 | ||
| 334 | 554 | if (seq.getAnnotation() != null) |
| 335 | { | |
| 336 | 11 | AlignmentAnnotation[] sqann = seq.getAnnotation(); |
| 337 | 22 | for (int i = 0; i < sqann.length; i++) |
| 338 | { | |
| 339 | 11 | if (sqann[i] == null) |
| 340 | { | |
| 341 | 0 | continue; |
| 342 | } | |
| 343 | 11 | boolean found = (alAnnotation == null); |
| 344 | 11 | if (!found) |
| 345 | { | |
| 346 | 22 | for (int apos = 0; !found && apos < alAnnotation.length; apos++) |
| 347 | { | |
| 348 | 11 | found = (alAnnotation[apos] == sqann[i]); |
| 349 | } | |
| 350 | } | |
| 351 | 11 | if (found) |
| 352 | { | |
| 353 | // only copy the given annotation | |
| 354 | 11 | AlignmentAnnotation newann = new AlignmentAnnotation(sqann[i]); |
| 355 | 11 | ContactMatrixI cm = seq.getContactMatrixFor(sqann[i]); |
| 356 | 11 | if (cm != null) |
| 357 | { | |
| 358 | 5 | addContactListFor(newann, cm); |
| 359 | } | |
| 360 | 11 | addAlignmentAnnotation(newann); |
| 361 | } | |
| 362 | } | |
| 363 | } | |
| 364 | 554 | if (seq.getAllPDBEntries() != null) |
| 365 | { | |
| 366 | 554 | Vector<PDBEntry> ids = seq.getAllPDBEntries(); |
| 367 | 554 | for (PDBEntry pdb : ids) |
| 368 | { | |
| 369 | 146 | this.addPDBId(new PDBEntry(pdb)); |
| 370 | } | |
| 371 | } | |
| 372 | } | |
| 373 | ||
| 374 | 44 | @Override |
| 375 | public void setSequenceFeatures(List<SequenceFeature> features) | |
| 376 | { | |
| 377 | 44 | if (datasetSequence != null) |
| 378 | { | |
| 379 | 1 | datasetSequence.setSequenceFeatures(features); |
| 380 | 1 | return; |
| 381 | } | |
| 382 | 43 | sequenceFeatureStore = new SequenceFeatures(features); |
| 383 | } | |
| 384 | ||
| 385 | 278629 | @Override |
| 386 | public synchronized boolean addSequenceFeature(SequenceFeature sf) | |
| 387 | { | |
| 388 | 278629 | if (sf.getType() == null) |
| 389 | { | |
| 390 | 1 | jalview.bin.Console.errPrintln( |
| 391 | "SequenceFeature type may not be null: " + sf.toString()); | |
| 392 | 1 | return false; |
| 393 | } | |
| 394 | ||
| 395 | 278628 | if (datasetSequence != null) |
| 396 | { | |
| 397 | 67063 | return datasetSequence.addSequenceFeature(sf); |
| 398 | } | |
| 399 | ||
| 400 | 211565 | return sequenceFeatureStore.add(sf); |
| 401 | } | |
| 402 | ||
| 403 | 308 | @Override |
| 404 | public void deleteFeature(SequenceFeature sf) | |
| 405 | { | |
| 406 | 308 | if (datasetSequence != null) |
| 407 | { | |
| 408 | 109 | datasetSequence.deleteFeature(sf); |
| 409 | } | |
| 410 | else | |
| 411 | { | |
| 412 | 199 | sequenceFeatureStore.delete(sf); |
| 413 | } | |
| 414 | } | |
| 415 | ||
| 416 | /** | |
| 417 | * {@inheritDoc} | |
| 418 | * | |
| 419 | * @return | |
| 420 | */ | |
| 421 | 8341 | @Override |
| 422 | public List<SequenceFeature> getSequenceFeatures() | |
| 423 | { | |
| 424 | 8341 | if (datasetSequence != null) |
| 425 | { | |
| 426 | 3175 | return datasetSequence.getSequenceFeatures(); |
| 427 | } | |
| 428 | 5166 | return sequenceFeatureStore.getAllFeatures(); |
| 429 | } | |
| 430 | ||
| 431 | 550163 | @Override |
| 432 | public SequenceFeaturesI getFeatures() | |
| 433 | { | |
| 434 | 550682 | return datasetSequence != null ? datasetSequence.getFeatures() |
| 435 | : sequenceFeatureStore; | |
| 436 | } | |
| 437 | ||
| 438 | 1221 | @Override |
| 439 | public boolean addPDBId(PDBEntry entry) | |
| 440 | { | |
| 441 | 1221 | if (pdbIds == null) |
| 442 | { | |
| 443 | 432 | pdbIds = new Vector<>(); |
| 444 | 432 | pdbIds.add(entry); |
| 445 | 432 | return true; |
| 446 | } | |
| 447 | ||
| 448 | 789 | for (PDBEntry pdbe : pdbIds) |
| 449 | { | |
| 450 | 17484 | if (pdbe.updateFrom(entry)) |
| 451 | { | |
| 452 | 457 | return false; |
| 453 | } | |
| 454 | } | |
| 455 | 332 | pdbIds.addElement(entry); |
| 456 | 332 | return true; |
| 457 | } | |
| 458 | ||
| 459 | /** | |
| 460 | * DOCUMENT ME! | |
| 461 | * | |
| 462 | * @param id | |
| 463 | * DOCUMENT ME! | |
| 464 | */ | |
| 465 | 21 | @Override |
| 466 | public void setPDBId(Vector<PDBEntry> id) | |
| 467 | { | |
| 468 | 21 | pdbIds = id; |
| 469 | } | |
| 470 | ||
| 471 | /** | |
| 472 | * DOCUMENT ME! | |
| 473 | * | |
| 474 | * @return DOCUMENT ME! | |
| 475 | */ | |
| 476 | 45833 | @Override |
| 477 | public Vector<PDBEntry> getAllPDBEntries() | |
| 478 | { | |
| 479 | 45833 | return pdbIds == null ? new Vector<>() : pdbIds; |
| 480 | } | |
| 481 | ||
| 482 | /** | |
| 483 | * Answers the sequence name, with '/start-end' appended if jvsuffix is true | |
| 484 | * | |
| 485 | * @return | |
| 486 | */ | |
| 487 | 53195 | @Override |
| 488 | public String getDisplayId(boolean jvsuffix) | |
| 489 | { | |
| 490 | 53195 | if (!jvsuffix) |
| 491 | { | |
| 492 | 714 | return name; |
| 493 | } | |
| 494 | 52481 | StringBuilder result = new StringBuilder(name); |
| 495 | 52481 | result.append("/").append(start).append("-").append(end); |
| 496 | ||
| 497 | 52481 | return result.toString(); |
| 498 | } | |
| 499 | ||
| 500 | /** | |
| 501 | * Sets the sequence name. If the name ends in /start-end, then the start-end | |
| 502 | * values are parsed out and set, and the suffix is removed from the name. | |
| 503 | * | |
| 504 | * @param theName | |
| 505 | */ | |
| 506 | 1056 | @Override |
| 507 | public void setName(String theName) | |
| 508 | { | |
| 509 | 1056 | this.name = theName; |
| 510 | 1056 | this.parseId(); |
| 511 | } | |
| 512 | ||
| 513 | /** | |
| 514 | * DOCUMENT ME! | |
| 515 | * | |
| 516 | * @return DOCUMENT ME! | |
| 517 | */ | |
| 518 | 55768 | @Override |
| 519 | public String getName() | |
| 520 | { | |
| 521 | 55768 | return this.name; |
| 522 | } | |
| 523 | ||
| 524 | /** | |
| 525 | * DOCUMENT ME! | |
| 526 | * | |
| 527 | * @param start | |
| 528 | * DOCUMENT ME! | |
| 529 | */ | |
| 530 | 2854 | @Override |
| 531 | public void setStart(int start) | |
| 532 | { | |
| 533 | 2854 | this.start = start; |
| 534 | 2854 | sequenceChanged(); |
| 535 | } | |
| 536 | ||
| 537 | /** | |
| 538 | * DOCUMENT ME! | |
| 539 | * | |
| 540 | * @return DOCUMENT ME! | |
| 541 | */ | |
| 542 | 226689 | @Override |
| 543 | public int getStart() | |
| 544 | { | |
| 545 | 226690 | return this.start; |
| 546 | } | |
| 547 | ||
| 548 | /** | |
| 549 | * DOCUMENT ME! | |
| 550 | * | |
| 551 | * @param end | |
| 552 | * DOCUMENT ME! | |
| 553 | */ | |
| 554 | 2851 | @Override |
| 555 | public void setEnd(int end) | |
| 556 | { | |
| 557 | 2851 | this.end = end; |
| 558 | } | |
| 559 | ||
| 560 | /** | |
| 561 | * DOCUMENT ME! | |
| 562 | * | |
| 563 | * @return DOCUMENT ME! | |
| 564 | */ | |
| 565 | 431808 | @Override |
| 566 | public int getEnd() | |
| 567 | { | |
| 568 | 431807 | return this.end; |
| 569 | } | |
| 570 | ||
| 571 | /** | |
| 572 | * DOCUMENT ME! | |
| 573 | * | |
| 574 | * @return DOCUMENT ME! | |
| 575 | */ | |
| 576 | 18359212 | @Override |
| 577 | public int getLength() | |
| 578 | { | |
| 579 | 18396394 | return this.sequence.length; |
| 580 | } | |
| 581 | ||
| 582 | /** | |
| 583 | * DOCUMENT ME! | |
| 584 | * | |
| 585 | * @param seq | |
| 586 | * DOCUMENT ME! | |
| 587 | */ | |
| 588 | 10552 | @Override |
| 589 | public void setSequence(String seq) | |
| 590 | { | |
| 591 | 10552 | this.sequence = seq.toCharArray(); |
| 592 | 10552 | checkValidRange(); |
| 593 | 10552 | sequenceChanged(); |
| 594 | } | |
| 595 | ||
| 596 | 28919 | @Override |
| 597 | public String getSequenceAsString() | |
| 598 | { | |
| 599 | 28919 | return new String(sequence); |
| 600 | } | |
| 601 | ||
| 602 | 19239 | @Override |
| 603 | public String getSequenceAsString(int start, int end) | |
| 604 | { | |
| 605 | 19239 | return new String(getSequence(start, end)); |
| 606 | } | |
| 607 | ||
| 608 | 16647 | @Override |
| 609 | public char[] getSequence() | |
| 610 | { | |
| 611 | // return sequence; | |
| 612 | 16647 | return sequence == null ? null |
| 613 | : Arrays.copyOf(sequence, sequence.length); | |
| 614 | } | |
| 615 | ||
| 616 | /* | |
| 617 | * (non-Javadoc) | |
| 618 | * | |
| 619 | * @see jalview.datamodel.SequenceI#getSequence(int, int) | |
| 620 | */ | |
| 621 | 21218 | @Override |
| 622 | public char[] getSequence(int start, int end) | |
| 623 | { | |
| 624 | 21218 | if (start < 0) |
| 625 | { | |
| 626 | 0 | start = 0; |
| 627 | } | |
| 628 | // JBPNote - left to user to pad the result here (TODO:Decide on this | |
| 629 | // policy) | |
| 630 | 21218 | if (start >= sequence.length) |
| 631 | { | |
| 632 | 2 | return new char[0]; |
| 633 | } | |
| 634 | ||
| 635 | 21216 | if (end >= sequence.length) |
| 636 | { | |
| 637 | 2420 | end = sequence.length; |
| 638 | } | |
| 639 | ||
| 640 | 21216 | char[] reply = new char[end - start]; |
| 641 | 21216 | System.arraycopy(sequence, start, reply, 0, end - start); |
| 642 | ||
| 643 | 21216 | return reply; |
| 644 | } | |
| 645 | ||
| 646 | 40 | @Override |
| 647 | public SequenceI getSubSequence(int start, int end) | |
| 648 | { | |
| 649 | 40 | if (start < 0) |
| 650 | { | |
| 651 | 0 | start = 0; |
| 652 | } | |
| 653 | 40 | char[] seq = getSequence(start, end); |
| 654 | 40 | if (seq.length == 0) |
| 655 | { | |
| 656 | 0 | return null; |
| 657 | } | |
| 658 | 40 | int nstart = findPosition(start); |
| 659 | 40 | int nend = findPosition(end) - 1; |
| 660 | // JBPNote - this is an incomplete copy. | |
| 661 | 40 | SequenceI nseq = new Sequence(this.getName(), seq, nstart, nend); |
| 662 | 40 | nseq.setDescription(description); |
| 663 | 40 | if (datasetSequence != null) |
| 664 | { | |
| 665 | 25 | nseq.setDatasetSequence(datasetSequence); |
| 666 | } | |
| 667 | else | |
| 668 | { | |
| 669 | 15 | nseq.setDatasetSequence(this); |
| 670 | } | |
| 671 | 40 | return nseq; |
| 672 | } | |
| 673 | ||
| 674 | /** | |
| 675 | * Returns the character of the aligned sequence at the given position (base | |
| 676 | * zero), or space if the position is not within the sequence's bounds | |
| 677 | * | |
| 678 | * @return | |
| 679 | */ | |
| 680 | 34204264 | @Override |
| 681 | public char getCharAt(int i) | |
| 682 | { | |
| 683 | 34635114 | if (i >= 0 && i < sequence.length) |
| 684 | { | |
| 685 | 35028999 | return sequence[i]; |
| 686 | } | |
| 687 | else | |
| 688 | { | |
| 689 | 37584 | return ' '; |
| 690 | } | |
| 691 | } | |
| 692 | ||
| 693 | /** | |
| 694 | * Sets the sequence description, and also parses out any special formats of | |
| 695 | * interest | |
| 696 | * | |
| 697 | * @param desc | |
| 698 | */ | |
| 699 | 9893 | @Override |
| 700 | public void setDescription(String desc) | |
| 701 | { | |
| 702 | 9893 | this.description = desc; |
| 703 | } | |
| 704 | ||
| 705 | 35 | @Override |
| 706 | public void setGeneLoci(String speciesId, String assemblyId, | |
| 707 | String chromosomeId, MapList map) | |
| 708 | { | |
| 709 | 35 | addDBRef(new GeneLocus(speciesId, assemblyId, chromosomeId, |
| 710 | new Mapping(map))); | |
| 711 | } | |
| 712 | ||
| 713 | /** | |
| 714 | * Returns the gene loci mapping for the sequence (may be null) | |
| 715 | * | |
| 716 | * @return | |
| 717 | */ | |
| 718 | 68 | @Override |
| 719 | public GeneLociI getGeneLoci() | |
| 720 | { | |
| 721 | 68 | List<DBRefEntry> refs = getDBRefs(); |
| 722 | 68 | if (refs != null) |
| 723 | { | |
| 724 | 50 | for (final DBRefEntry ref : refs) |
| 725 | { | |
| 726 | 63 | if (ref instanceof GeneLociI) |
| 727 | { | |
| 728 | 35 | return (GeneLociI) ref; |
| 729 | } | |
| 730 | } | |
| 731 | } | |
| 732 | 33 | return null; |
| 733 | } | |
| 734 | ||
| 735 | /** | |
| 736 | * Answers the description | |
| 737 | * | |
| 738 | * @return | |
| 739 | */ | |
| 740 | 4767 | @Override |
| 741 | public String getDescription() | |
| 742 | { | |
| 743 | 4767 | return this.description; |
| 744 | } | |
| 745 | ||
| 746 | /** | |
| 747 | * {@inheritDoc} | |
| 748 | */ | |
| 749 | 500764 | @Override |
| 750 | public int findIndex(int pos) | |
| 751 | { | |
| 752 | /* | |
| 753 | * use a valid, hopefully nearby, cursor if available | |
| 754 | */ | |
| 755 | 500783 | if (isValidCursor(cursor)) |
| 756 | { | |
| 757 | 500106 | return findIndex(pos, cursor); |
| 758 | } | |
| 759 | ||
| 760 | 681 | int j = start; |
| 761 | 681 | int i = 0; |
| 762 | 681 | int startColumn = 0; |
| 763 | ||
| 764 | /* | |
| 765 | * traverse sequence from the start counting gaps; make a note of | |
| 766 | * the column of the first residue to save in the cursor | |
| 767 | */ | |
| 768 | 447342 | while ((i < sequence.length) && (j <= end) && (j <= pos)) |
| 769 | { | |
| 770 | 446661 | if (!Comparison.isGap(sequence[i])) |
| 771 | { | |
| 772 | 52026 | if (j == start) |
| 773 | { | |
| 774 | 675 | startColumn = i; |
| 775 | } | |
| 776 | 52026 | j++; |
| 777 | } | |
| 778 | 446661 | i++; |
| 779 | } | |
| 780 | ||
| 781 | 681 | if (j == end && j < pos) |
| 782 | { | |
| 783 | 0 | return end + 1; |
| 784 | } | |
| 785 | ||
| 786 | 681 | updateCursor(pos, i, startColumn); |
| 787 | 681 | return i; |
| 788 | } | |
| 789 | ||
| 790 | /** | |
| 791 | * Updates the cursor to the latest found residue and column position | |
| 792 | * | |
| 793 | * @param residuePos | |
| 794 | * (start..) | |
| 795 | * @param column | |
| 796 | * (1..) | |
| 797 | * @param startColumn | |
| 798 | * column position of the first sequence residue | |
| 799 | */ | |
| 800 | 763022 | protected void updateCursor(int residuePos, int column, int startColumn) |
| 801 | { | |
| 802 | /* | |
| 803 | * preserve end residue column provided cursor was valid | |
| 804 | */ | |
| 805 | 763027 | int endColumn = isValidCursor(cursor) ? cursor.lastColumnPosition : 0; |
| 806 | ||
| 807 | 763032 | if (residuePos == this.end) |
| 808 | { | |
| 809 | 7509 | endColumn = column; |
| 810 | } | |
| 811 | ||
| 812 | 763032 | cursor = new SequenceCursor(this, residuePos, column, startColumn, |
| 813 | endColumn, this.changeCount); | |
| 814 | } | |
| 815 | ||
| 816 | /** | |
| 817 | * Answers the aligned column position (1..) for the given residue position | |
| 818 | * (start..) given a 'hint' of a residue/column location in the neighbourhood. | |
| 819 | * The hint may be left of, at, or to the right of the required position. | |
| 820 | * | |
| 821 | * @param pos | |
| 822 | * @param curs | |
| 823 | * @return | |
| 824 | */ | |
| 825 | 500110 | protected int findIndex(final int pos, SequenceCursor curs) |
| 826 | { | |
| 827 | 500111 | if (!isValidCursor(curs)) |
| 828 | { | |
| 829 | /* | |
| 830 | * wrong or invalidated cursor, compute de novo | |
| 831 | */ | |
| 832 | 0 | return findIndex(pos); |
| 833 | } | |
| 834 | ||
| 835 | 500112 | if (curs.residuePosition == pos) |
| 836 | { | |
| 837 | 22358 | return curs.columnPosition; |
| 838 | } | |
| 839 | ||
| 840 | /* | |
| 841 | * move left or right to find pos from hint.position | |
| 842 | */ | |
| 843 | 477754 | int col = curs.columnPosition - 1; // convert from base 1 to base 0 |
| 844 | 477754 | int newPos = curs.residuePosition; |
| 845 | 477769 | int delta = newPos > pos ? -1 : 1; |
| 846 | ||
| 847 | 2165786 | while (newPos != pos) |
| 848 | { | |
| 849 | 1688063 | col += delta; // shift one column left or right |
| 850 | 1688063 | if (col < 0) |
| 851 | { | |
| 852 | 11 | break; |
| 853 | } | |
| 854 | 1688056 | if (col == sequence.length) |
| 855 | { | |
| 856 | 46 | col--; // return last column if we failed to reach pos |
| 857 | 46 | break; |
| 858 | } | |
| 859 | 1688016 | if (!Comparison.isGap(sequence[col])) |
| 860 | { | |
| 861 | 1249072 | newPos += delta; |
| 862 | } | |
| 863 | } | |
| 864 | ||
| 865 | 477780 | col++; // convert back to base 1 |
| 866 | ||
| 867 | /* | |
| 868 | * only update cursor if we found the target position | |
| 869 | */ | |
| 870 | 477780 | if (newPos == pos) |
| 871 | { | |
| 872 | 477723 | updateCursor(pos, col, curs.firstColumnPosition); |
| 873 | } | |
| 874 | ||
| 875 | 477793 | return col; |
| 876 | } | |
| 877 | ||
| 878 | /** | |
| 879 | * {@inheritDoc} | |
| 880 | */ | |
| 881 | 530658 | @Override |
| 882 | public int findPosition(final int column) | |
| 883 | { | |
| 884 | /* | |
| 885 | * use a valid, hopefully nearby, cursor if available | |
| 886 | */ | |
| 887 | 530740 | if (isValidCursor(cursor)) |
| 888 | { | |
| 889 | 528694 | return findPosition(column + 1, cursor); |
| 890 | } | |
| 891 | ||
| 892 | // TODO recode this more naturally i.e. count residues only | |
| 893 | // as they are found, not 'in anticipation' | |
| 894 | ||
| 895 | /* | |
| 896 | * traverse the sequence counting gaps; note the column position | |
| 897 | * of the first residue, to save in the cursor | |
| 898 | */ | |
| 899 | 2058 | int firstResidueColumn = 0; |
| 900 | 2058 | int lastPosFound = 0; |
| 901 | 2058 | int lastPosFoundColumn = 0; |
| 902 | 2058 | int seqlen = sequence.length; |
| 903 | ||
| 904 | 2058 | if (seqlen > 0 && !Comparison.isGap(sequence[0])) |
| 905 | { | |
| 906 | 1690 | lastPosFound = start; |
| 907 | 1690 | lastPosFoundColumn = 0; |
| 908 | } | |
| 909 | ||
| 910 | 2058 | int j = 0; |
| 911 | 2058 | int pos = start; |
| 912 | ||
| 913 | 44407 | while (j < column && j < seqlen) |
| 914 | { | |
| 915 | 42349 | if (!Comparison.isGap(sequence[j])) |
| 916 | { | |
| 917 | 33192 | lastPosFound = pos; |
| 918 | 33192 | lastPosFoundColumn = j; |
| 919 | 33192 | if (pos == this.start) |
| 920 | { | |
| 921 | 603 | firstResidueColumn = j; |
| 922 | } | |
| 923 | 33192 | pos++; |
| 924 | } | |
| 925 | 42349 | j++; |
| 926 | } | |
| 927 | 2058 | if (j < seqlen && !Comparison.isGap(sequence[j])) |
| 928 | { | |
| 929 | 1833 | lastPosFound = pos; |
| 930 | 1833 | lastPosFoundColumn = j; |
| 931 | 1833 | if (pos == this.start) |
| 932 | { | |
| 933 | 1399 | firstResidueColumn = j; |
| 934 | } | |
| 935 | } | |
| 936 | ||
| 937 | /* | |
| 938 | * update the cursor to the last residue position found (if any) | |
| 939 | * (converting column position to base 1) | |
| 940 | */ | |
| 941 | 2058 | if (lastPosFound != 0) |
| 942 | { | |
| 943 | 2002 | updateCursor(lastPosFound, lastPosFoundColumn + 1, |
| 944 | firstResidueColumn + 1); | |
| 945 | } | |
| 946 | ||
| 947 | 2058 | return pos; |
| 948 | } | |
| 949 | ||
| 950 | /** | |
| 951 | * Answers true if the given cursor is not null, is for this sequence object, | |
| 952 | * and has a token value that matches this object's changeCount, else false. | |
| 953 | * This allows us to ignore a cursor as 'stale' if the sequence has been | |
| 954 | * modified since the cursor was created. | |
| 955 | * | |
| 956 | * @param curs | |
| 957 | * @return | |
| 958 | */ | |
| 959 | 2822098 | protected boolean isValidCursor(SequenceCursor curs) |
| 960 | { | |
| 961 | 2822116 | if (curs == null || curs.sequence != this || curs.token != changeCount) |
| 962 | { | |
| 963 | 5427 | return false; |
| 964 | } | |
| 965 | /* | |
| 966 | * sanity check against range | |
| 967 | */ | |
| 968 | 2817118 | if (curs.columnPosition < 0 || curs.columnPosition > sequence.length) |
| 969 | { | |
| 970 | 0 | return false; |
| 971 | } | |
| 972 | 2817433 | if (curs.residuePosition < start || curs.residuePosition > end) |
| 973 | { | |
| 974 | 0 | return false; |
| 975 | } | |
| 976 | 2817415 | return true; |
| 977 | } | |
| 978 | ||
| 979 | /** | |
| 980 | * Answers the sequence position (start..) for the given aligned column | |
| 981 | * position (1..), given a hint of a cursor in the neighbourhood. The cursor | |
| 982 | * may lie left of, at, or to the right of the column position. | |
| 983 | * | |
| 984 | * @param col | |
| 985 | * @param curs | |
| 986 | * @return | |
| 987 | */ | |
| 988 | 528677 | protected int findPosition(final int col, SequenceCursor curs) |
| 989 | { | |
| 990 | 528707 | if (!isValidCursor(curs)) |
| 991 | { | |
| 992 | /* | |
| 993 | * wrong or invalidated cursor, compute de novo | |
| 994 | */ | |
| 995 | 2 | return findPosition(col - 1);// ugh back to base 0 |
| 996 | } | |
| 997 | ||
| 998 | 528717 | if (curs.columnPosition == col) |
| 999 | { | |
| 1000 | 238253 | cursor = curs; // in case this method becomes public |
| 1001 | 238224 | return curs.residuePosition; // easy case :-) |
| 1002 | } | |
| 1003 | ||
| 1004 | 290480 | if (curs.lastColumnPosition > 0 && curs.lastColumnPosition < col) |
| 1005 | { | |
| 1006 | /* | |
| 1007 | * sequence lies entirely to the left of col | |
| 1008 | * - return last residue + 1 | |
| 1009 | */ | |
| 1010 | 197 | return end + 1; |
| 1011 | } | |
| 1012 | ||
| 1013 | 290285 | if (curs.firstColumnPosition > 0 && curs.firstColumnPosition > col) |
| 1014 | { | |
| 1015 | /* | |
| 1016 | * sequence lies entirely to the right of col | |
| 1017 | * - return first residue | |
| 1018 | */ | |
| 1019 | 97 | return start; |
| 1020 | } | |
| 1021 | ||
| 1022 | // todo could choose closest to col out of column, | |
| 1023 | // firstColumnPosition, lastColumnPosition as a start point | |
| 1024 | ||
| 1025 | /* | |
| 1026 | * move left or right to find pos from cursor position | |
| 1027 | */ | |
| 1028 | 290189 | int firstResidueColumn = curs.firstColumnPosition; |
| 1029 | 290190 | int column = curs.columnPosition - 1; // to base 0 |
| 1030 | 290192 | int newPos = curs.residuePosition; |
| 1031 | 290191 | int delta = curs.columnPosition > col ? -1 : 1; |
| 1032 | 290191 | boolean gapped = false; |
| 1033 | 290189 | int lastFoundPosition = curs.residuePosition; |
| 1034 | 290188 | int lastFoundPositionColumn = curs.columnPosition; |
| 1035 | ||
| 1036 | 1798394 | while (column != col - 1) |
| 1037 | { | |
| 1038 | 1508411 | column += delta; // shift one column left or right |
| 1039 | 1508845 | if (column < 0 || column == sequence.length) |
| 1040 | { | |
| 1041 | 117 | break; |
| 1042 | } | |
| 1043 | 1508936 | gapped = Comparison.isGap(sequence[column]); |
| 1044 | 1508713 | if (!gapped) |
| 1045 | { | |
| 1046 | 1032096 | newPos += delta; |
| 1047 | 1032095 | lastFoundPosition = newPos; |
| 1048 | 1032143 | lastFoundPositionColumn = column + 1; |
| 1049 | 1032473 | if (lastFoundPosition == this.start) |
| 1050 | { | |
| 1051 | 1652 | firstResidueColumn = column + 1; |
| 1052 | } | |
| 1053 | } | |
| 1054 | } | |
| 1055 | ||
| 1056 | 290186 | if (cursor == null || lastFoundPosition != cursor.residuePosition) |
| 1057 | { | |
| 1058 | 282634 | updateCursor(lastFoundPosition, lastFoundPositionColumn, |
| 1059 | firstResidueColumn); | |
| 1060 | } | |
| 1061 | ||
| 1062 | /* | |
| 1063 | * hack to give position to the right if on a gap | |
| 1064 | * or beyond the length of the sequence (see JAL-2562) | |
| 1065 | */ | |
| 1066 | 290195 | if (delta > 0 && (gapped || column >= sequence.length)) |
| 1067 | { | |
| 1068 | 7690 | newPos++; |
| 1069 | } | |
| 1070 | ||
| 1071 | 290192 | return newPos; |
| 1072 | } | |
| 1073 | ||
| 1074 | /** | |
| 1075 | * {@inheritDoc} | |
| 1076 | */ | |
| 1077 | 1218 | @Override |
| 1078 | public ContiguousI findPositions(int fromColumn, int toColumn) | |
| 1079 | { | |
| 1080 | 1218 | if (toColumn < fromColumn || fromColumn < 1) |
| 1081 | { | |
| 1082 | 10 | return null; |
| 1083 | } | |
| 1084 | ||
| 1085 | /* | |
| 1086 | * find the first non-gapped position, if any | |
| 1087 | */ | |
| 1088 | 1208 | int firstPosition = 0; |
| 1089 | 1208 | int col = fromColumn - 1; |
| 1090 | 1208 | int length = sequence.length; |
| 1091 | 11144 | while (col < length && col < toColumn) |
| 1092 | { | |
| 1093 | 10964 | if (!Comparison.isGap(sequence[col])) |
| 1094 | { | |
| 1095 | 1028 | firstPosition = findPosition(col++); |
| 1096 | 1028 | break; |
| 1097 | } | |
| 1098 | 9936 | col++; |
| 1099 | } | |
| 1100 | ||
| 1101 | 1208 | if (firstPosition == 0) |
| 1102 | { | |
| 1103 | 180 | return null; |
| 1104 | } | |
| 1105 | ||
| 1106 | /* | |
| 1107 | * find the last non-gapped position | |
| 1108 | */ | |
| 1109 | 1028 | int lastPosition = firstPosition; |
| 1110 | 43176 | while (col < length && col < toColumn) |
| 1111 | { | |
| 1112 | 42148 | if (!Comparison.isGap(sequence[col++])) |
| 1113 | { | |
| 1114 | 42105 | lastPosition++; |
| 1115 | } | |
| 1116 | } | |
| 1117 | ||
| 1118 | 1028 | return new Range(firstPosition, lastPosition); |
| 1119 | } | |
| 1120 | ||
| 1121 | /** | |
| 1122 | * Returns an int array where indices correspond to each residue in the | |
| 1123 | * sequence and the element value gives its position in the alignment | |
| 1124 | * | |
| 1125 | * @return int[SequenceI.getEnd()-SequenceI.getStart()+1] or null if no | |
| 1126 | * residues in SequenceI object | |
| 1127 | */ | |
| 1128 | 1 | @Override |
| 1129 | public int[] gapMap() | |
| 1130 | { | |
| 1131 | 1 | String seq = jalview.analysis.AlignSeq.extractGaps( |
| 1132 | jalview.util.Comparison.GapChars, new String(sequence)); | |
| 1133 | 1 | int[] map = new int[seq.length()]; |
| 1134 | 1 | int j = 0; |
| 1135 | 1 | int p = 0; |
| 1136 | ||
| 1137 | 15 | while (j < sequence.length) |
| 1138 | { | |
| 1139 | 14 | if (!jalview.util.Comparison.isGap(sequence[j])) |
| 1140 | { | |
| 1141 | 6 | map[p++] = j; |
| 1142 | } | |
| 1143 | ||
| 1144 | 14 | j++; |
| 1145 | } | |
| 1146 | ||
| 1147 | 1 | return map; |
| 1148 | } | |
| 1149 | ||
| 1150 | /** | |
| 1151 | * Build a bitset corresponding to sequence gaps | |
| 1152 | * | |
| 1153 | * @return a BitSet where set values correspond to gaps in the sequence | |
| 1154 | */ | |
| 1155 | 3 | @Override |
| 1156 | public BitSet gapBitset() | |
| 1157 | { | |
| 1158 | 3 | BitSet gaps = new BitSet(sequence.length); |
| 1159 | 3 | int j = 0; |
| 1160 | 69 | while (j < sequence.length) |
| 1161 | { | |
| 1162 | 66 | if (jalview.util.Comparison.isGap(sequence[j])) |
| 1163 | { | |
| 1164 | 15 | gaps.set(j); |
| 1165 | } | |
| 1166 | 66 | j++; |
| 1167 | } | |
| 1168 | 3 | return gaps; |
| 1169 | } | |
| 1170 | ||
| 1171 | 321 | @Override |
| 1172 | public int[] findPositionMap() | |
| 1173 | { | |
| 1174 | 321 | int map[] = new int[sequence.length]; |
| 1175 | 321 | int j = 0; |
| 1176 | 321 | int pos = start; |
| 1177 | 321 | int seqlen = sequence.length; |
| 1178 | 24663 | while ((j < seqlen)) |
| 1179 | { | |
| 1180 | 24342 | map[j] = pos; |
| 1181 | 24342 | if (!jalview.util.Comparison.isGap(sequence[j])) |
| 1182 | { | |
| 1183 | 16801 | pos++; |
| 1184 | } | |
| 1185 | ||
| 1186 | 24342 | j++; |
| 1187 | } | |
| 1188 | 321 | return map; |
| 1189 | } | |
| 1190 | ||
| 1191 | 4 | @Override |
| 1192 | public List<int[]> getInsertions() | |
| 1193 | { | |
| 1194 | 4 | ArrayList<int[]> map = new ArrayList<>(); |
| 1195 | 4 | int lastj = -1, j = 0; |
| 1196 | // int pos = start; | |
| 1197 | 4 | int seqlen = sequence.length; |
| 1198 | 220 | while ((j < seqlen)) |
| 1199 | { | |
| 1200 | 216 | if (jalview.util.Comparison.isGap(sequence[j])) |
| 1201 | { | |
| 1202 | 25 | if (lastj == -1) |
| 1203 | { | |
| 1204 | 11 | lastj = j; |
| 1205 | } | |
| 1206 | } | |
| 1207 | else | |
| 1208 | { | |
| 1209 | 191 | if (lastj != -1) |
| 1210 | { | |
| 1211 | 10 | map.add(new int[] { lastj, j - 1 }); |
| 1212 | 10 | lastj = -1; |
| 1213 | } | |
| 1214 | } | |
| 1215 | 216 | j++; |
| 1216 | } | |
| 1217 | 4 | if (lastj != -1) |
| 1218 | { | |
| 1219 | 1 | map.add(new int[] { lastj, j - 1 }); |
| 1220 | 1 | lastj = -1; |
| 1221 | } | |
| 1222 | 4 | return map; |
| 1223 | } | |
| 1224 | ||
| 1225 | 5 | @Override |
| 1226 | public BitSet getInsertionsAsBits() | |
| 1227 | { | |
| 1228 | 5 | BitSet map = new BitSet(); |
| 1229 | 5 | int lastj = -1, j = 0; |
| 1230 | // int pos = start; | |
| 1231 | 5 | int seqlen = sequence.length; |
| 1232 | 187 | while ((j < seqlen)) |
| 1233 | { | |
| 1234 | 182 | if (jalview.util.Comparison.isGap(sequence[j])) |
| 1235 | { | |
| 1236 | 43 | if (lastj == -1) |
| 1237 | { | |
| 1238 | 16 | lastj = j; |
| 1239 | } | |
| 1240 | } | |
| 1241 | else | |
| 1242 | { | |
| 1243 | 139 | if (lastj != -1) |
| 1244 | { | |
| 1245 | 15 | map.set(lastj, j); |
| 1246 | 15 | lastj = -1; |
| 1247 | } | |
| 1248 | } | |
| 1249 | 182 | j++; |
| 1250 | } | |
| 1251 | 5 | if (lastj != -1) |
| 1252 | { | |
| 1253 | 1 | map.set(lastj, j); |
| 1254 | 1 | lastj = -1; |
| 1255 | } | |
| 1256 | 5 | return map; |
| 1257 | } | |
| 1258 | ||
| 1259 | 175 | @Override |
| 1260 | public void deleteChars(final int i, final int j) | |
| 1261 | { | |
| 1262 | 175 | int newstart = start, newend = end; |
| 1263 | 175 | if (i >= sequence.length || i < 0) |
| 1264 | { | |
| 1265 | 3 | return; |
| 1266 | } | |
| 1267 | ||
| 1268 | 172 | char[] tmp = StringUtils.deleteChars(sequence, i, j); |
| 1269 | 172 | boolean createNewDs = false; |
| 1270 | // TODO: take a (second look) at the dataset creation validation method for | |
| 1271 | // the very large sequence case | |
| 1272 | ||
| 1273 | 172 | int startIndex = findIndex(start) - 1; |
| 1274 | 172 | int endIndex = findIndex(end) - 1; |
| 1275 | 172 | int startDeleteColumn = -1; // for dataset sequence deletions |
| 1276 | 172 | int deleteCount = 0; |
| 1277 | ||
| 1278 | 527 | for (int s = i; s < j && s < sequence.length; s++) |
| 1279 | { | |
| 1280 | 397 | if (Comparison.isGap(sequence[s])) |
| 1281 | { | |
| 1282 | 30 | continue; |
| 1283 | } | |
| 1284 | 367 | deleteCount++; |
| 1285 | 367 | if (startDeleteColumn == -1) |
| 1286 | { | |
| 1287 | 154 | startDeleteColumn = findPosition(s) - start; |
| 1288 | } | |
| 1289 | 367 | if (createNewDs) |
| 1290 | { | |
| 1291 | 213 | newend--; |
| 1292 | } | |
| 1293 | else | |
| 1294 | { | |
| 1295 | 154 | if (startIndex == s) |
| 1296 | { | |
| 1297 | /* | |
| 1298 | * deleting characters from start of sequence; new start is the | |
| 1299 | * sequence position of the next column (position to the right | |
| 1300 | * if the column position is gapped) | |
| 1301 | */ | |
| 1302 | 21 | newstart = findPosition(j); |
| 1303 | 21 | break; |
| 1304 | } | |
| 1305 | else | |
| 1306 | { | |
| 1307 | 133 | if (endIndex < j) |
| 1308 | { | |
| 1309 | /* | |
| 1310 | * deleting characters at end of sequence; new end is the sequence | |
| 1311 | * position of the column before the deletion; subtract 1 if this is | |
| 1312 | * gapped since findPosition returns the next sequence position | |
| 1313 | */ | |
| 1314 | 21 | newend = findPosition(i - 1); |
| 1315 | 21 | if (Comparison.isGap(sequence[i - 1])) |
| 1316 | { | |
| 1317 | 1 | newend--; |
| 1318 | } | |
| 1319 | 21 | break; |
| 1320 | } | |
| 1321 | else | |
| 1322 | { | |
| 1323 | 112 | createNewDs = true; |
| 1324 | 112 | newend--; |
| 1325 | } | |
| 1326 | } | |
| 1327 | } | |
| 1328 | } | |
| 1329 | ||
| 1330 | 172 | if (createNewDs && this.datasetSequence != null) |
| 1331 | { | |
| 1332 | /* | |
| 1333 | * if deletion occured in the middle of the sequence, | |
| 1334 | * construct a new dataset sequence and delete the residues | |
| 1335 | * that were deleted from the aligned sequence | |
| 1336 | */ | |
| 1337 | 55 | Sequence ds = new Sequence(datasetSequence); |
| 1338 | 55 | ds.deleteChars(startDeleteColumn, startDeleteColumn + deleteCount); |
| 1339 | 55 | datasetSequence = ds; |
| 1340 | // TODO: remove any non-inheritable properties ? | |
| 1341 | // TODO: create a sequence mapping (since there is a relation here ?) | |
| 1342 | } | |
| 1343 | 172 | start = newstart; |
| 1344 | 172 | end = newend; |
| 1345 | 172 | sequence = tmp; |
| 1346 | 172 | sequenceChanged(); |
| 1347 | } | |
| 1348 | ||
| 1349 | 732 | @Override |
| 1350 | public void insertCharAt(int i, int length, char c) | |
| 1351 | { | |
| 1352 | 732 | char[] tmp = new char[sequence.length + length]; |
| 1353 | ||
| 1354 | 732 | if (i >= sequence.length) |
| 1355 | { | |
| 1356 | 79 | System.arraycopy(sequence, 0, tmp, 0, sequence.length); |
| 1357 | 79 | i = sequence.length; |
| 1358 | } | |
| 1359 | else | |
| 1360 | { | |
| 1361 | 653 | System.arraycopy(sequence, 0, tmp, 0, i); |
| 1362 | } | |
| 1363 | ||
| 1364 | 732 | int index = i; |
| 1365 | 5432 | while (length > 0) |
| 1366 | { | |
| 1367 | 4700 | tmp[index++] = c; |
| 1368 | 4700 | length--; |
| 1369 | } | |
| 1370 | ||
| 1371 | 732 | if (i < sequence.length) |
| 1372 | { | |
| 1373 | 653 | System.arraycopy(sequence, i, tmp, index, sequence.length - i); |
| 1374 | } | |
| 1375 | ||
| 1376 | 732 | sequence = tmp; |
| 1377 | 732 | sequenceChanged(); |
| 1378 | } | |
| 1379 | ||
| 1380 | 608 | @Override |
| 1381 | public void insertCharAt(int i, char c) | |
| 1382 | { | |
| 1383 | 608 | insertCharAt(i, 1, c); |
| 1384 | } | |
| 1385 | ||
| 1386 | 0 | @Override |
| 1387 | public String getVamsasId() | |
| 1388 | { | |
| 1389 | 0 | return vamsasId; |
| 1390 | } | |
| 1391 | ||
| 1392 | 742 | @Override |
| 1393 | public void setVamsasId(String id) | |
| 1394 | { | |
| 1395 | 742 | vamsasId = id; |
| 1396 | } | |
| 1397 | ||
| 1398 | 8 | @Deprecated |
| 1399 | @Override | |
| 1400 | public void setDBRefs(DBModList<DBRefEntry> newDBrefs) | |
| 1401 | { | |
| 1402 | 8 | if (dbrefs == null && datasetSequence != null |
| 1403 | && this != datasetSequence) | |
| 1404 | { | |
| 1405 | 2 | datasetSequence.setDBRefs(newDBrefs); |
| 1406 | 2 | return; |
| 1407 | } | |
| 1408 | 6 | dbrefs = newDBrefs; |
| 1409 | 6 | refModCount = 0; |
| 1410 | } | |
| 1411 | ||
| 1412 | 40117 | @Override |
| 1413 | public DBModList<DBRefEntry> getDBRefs() | |
| 1414 | { | |
| 1415 | 40117 | if (dbrefs == null && datasetSequence != null |
| 1416 | && this != datasetSequence) | |
| 1417 | { | |
| 1418 | 14615 | return datasetSequence.getDBRefs(); |
| 1419 | } | |
| 1420 | 25502 | return dbrefs; |
| 1421 | } | |
| 1422 | ||
| 1423 | 3734 | @Override |
| 1424 | public void addDBRef(DBRefEntry entry) | |
| 1425 | { | |
| 1426 | // TODO JAL-3980 maintain as sorted list | |
| 1427 | 3734 | if (datasetSequence != null) |
| 1428 | { | |
| 1429 | 196 | datasetSequence.addDBRef(entry); |
| 1430 | 196 | return; |
| 1431 | } | |
| 1432 | ||
| 1433 | 3538 | if (dbrefs == null) |
| 1434 | { | |
| 1435 | 1639 | dbrefs = new DBModList<>(); |
| 1436 | } | |
| 1437 | // TODO JAL-3979 LOOK UP RATHER THAN SWEEP FOR EFFICIENCY | |
| 1438 | ||
| 1439 | 71780 | for (int ib = 0, nb = dbrefs.size(); ib < nb; ib++) |
| 1440 | { | |
| 1441 | 68281 | if (dbrefs.get(ib).updateFrom(entry)) |
| 1442 | { | |
| 1443 | /* | |
| 1444 | * found a dbref that either matched, or could be | |
| 1445 | * updated from, the new entry - no need to add it | |
| 1446 | */ | |
| 1447 | 39 | return; |
| 1448 | } | |
| 1449 | } | |
| 1450 | ||
| 1451 | // /// BH OUCH! | |
| 1452 | // /* | |
| 1453 | // * extend the array to make room for one more | |
| 1454 | // */ | |
| 1455 | // // TODO use an ArrayList instead | |
| 1456 | // int j = dbrefs.length; | |
| 1457 | // List<DBRefEntry> temp = new DBRefEntry[j + 1]; | |
| 1458 | // System.arraycopy(dbrefs, 0, temp, 0, j); | |
| 1459 | // temp[temp.length - 1] = entry; | |
| 1460 | // | |
| 1461 | // dbrefs = temp; | |
| 1462 | ||
| 1463 | 3499 | dbrefs.add(entry); |
| 1464 | } | |
| 1465 | ||
| 1466 | 1576 | @Override |
| 1467 | public void setDatasetSequence(SequenceI seq) | |
| 1468 | { | |
| 1469 | 1576 | if (seq == this) |
| 1470 | { | |
| 1471 | 1 | throw new IllegalArgumentException( |
| 1472 | "Implementation Error: self reference passed to SequenceI.setDatasetSequence"); | |
| 1473 | } | |
| 1474 | 1575 | if (seq != null && seq.getDatasetSequence() != null) |
| 1475 | { | |
| 1476 | 2 | throw new IllegalArgumentException( |
| 1477 | "Implementation error: cascading dataset sequences are not allowed."); | |
| 1478 | } | |
| 1479 | 1573 | datasetSequence = seq; |
| 1480 | } | |
| 1481 | ||
| 1482 | 731936 | @Override |
| 1483 | public SequenceI getDatasetSequence() | |
| 1484 | { | |
| 1485 | 732861 | return datasetSequence; |
| 1486 | } | |
| 1487 | ||
| 1488 | 9368 | @Override |
| 1489 | public AlignmentAnnotation[] getAnnotation() | |
| 1490 | { | |
| 1491 | 9368 | return annotation == null ? null |
| 1492 | : annotation | |
| 1493 | .toArray(new AlignmentAnnotation[annotation.size()]); | |
| 1494 | } | |
| 1495 | ||
| 1496 | 99 | @Override |
| 1497 | public boolean hasAnnotation(AlignmentAnnotation ann) | |
| 1498 | { | |
| 1499 | 99 | return annotation == null ? false : annotation.contains(ann); |
| 1500 | } | |
| 1501 | ||
| 1502 | 2014 | @Override |
| 1503 | public void addAlignmentAnnotation(AlignmentAnnotation annotation) | |
| 1504 | { | |
| 1505 | 2014 | if (this.annotation == null) |
| 1506 | { | |
| 1507 | 1374 | this.annotation = new Vector<>(); |
| 1508 | } | |
| 1509 | 2014 | if (!this.annotation.contains(annotation)) |
| 1510 | { | |
| 1511 | 2013 | this.annotation.addElement(annotation); |
| 1512 | } | |
| 1513 | 2014 | annotation.setSequenceRef(this); |
| 1514 | } | |
| 1515 | ||
| 1516 | 8 | @Override |
| 1517 | public void removeAlignmentAnnotation(AlignmentAnnotation annotation) | |
| 1518 | { | |
| 1519 | 8 | if (this.annotation != null) |
| 1520 | { | |
| 1521 | 8 | this.annotation.removeElement(annotation); |
| 1522 | 8 | if (this.annotation.size() == 0) |
| 1523 | { | |
| 1524 | 8 | this.annotation = null; |
| 1525 | } | |
| 1526 | } | |
| 1527 | } | |
| 1528 | ||
| 1529 | /** | |
| 1530 | * test if this is a valid candidate for another sequence's dataset sequence. | |
| 1531 | * | |
| 1532 | */ | |
| 1533 | 297 | private boolean isValidDatasetSequence() |
| 1534 | { | |
| 1535 | 297 | if (datasetSequence != null) |
| 1536 | { | |
| 1537 | 0 | return false; |
| 1538 | } | |
| 1539 | 66833 | for (int i = 0; i < sequence.length; i++) |
| 1540 | { | |
| 1541 | 66567 | if (jalview.util.Comparison.isGap(sequence[i])) |
| 1542 | { | |
| 1543 | 31 | return false; |
| 1544 | } | |
| 1545 | } | |
| 1546 | 266 | return true; |
| 1547 | } | |
| 1548 | ||
| 1549 | 430 | @Override |
| 1550 | public SequenceI deriveSequence() | |
| 1551 | { | |
| 1552 | 430 | Sequence seq = null; |
| 1553 | 430 | if (datasetSequence == null) |
| 1554 | { | |
| 1555 | 297 | if (isValidDatasetSequence()) |
| 1556 | { | |
| 1557 | // Use this as dataset sequence | |
| 1558 | 266 | seq = new Sequence(getName(), "", 1, -1); |
| 1559 | 266 | seq.setDatasetSequence(this); |
| 1560 | 266 | seq.initSeqFrom(this, getAnnotation()); |
| 1561 | 266 | return seq; |
| 1562 | } | |
| 1563 | else | |
| 1564 | { | |
| 1565 | // Create a new, valid dataset sequence | |
| 1566 | 31 | createDatasetSequence(); |
| 1567 | } | |
| 1568 | } | |
| 1569 | 164 | return new Sequence(this); |
| 1570 | } | |
| 1571 | ||
| 1572 | private boolean _isNa; | |
| 1573 | ||
| 1574 | private int _seqhash = 0; | |
| 1575 | ||
| 1576 | private List<DBRefEntry> primaryRefs; | |
| 1577 | ||
| 1578 | /** | |
| 1579 | * Answers false if the sequence is more than 85% nucleotide (ACGTU), else | |
| 1580 | * true | |
| 1581 | */ | |
| 1582 | 45845 | @Override |
| 1583 | public boolean isProtein() | |
| 1584 | { | |
| 1585 | 45845 | if (datasetSequence != null) |
| 1586 | { | |
| 1587 | 916 | return datasetSequence.isProtein(); |
| 1588 | } | |
| 1589 | 44929 | if (_seqhash != sequence.hashCode()) |
| 1590 | { | |
| 1591 | 2420 | _seqhash = sequence.hashCode(); |
| 1592 | 2420 | _isNa = Comparison.isNucleotide(this); |
| 1593 | } | |
| 1594 | 44929 | return !_isNa; |
| 1595 | } | |
| 1596 | ||
| 1597 | /* | |
| 1598 | * (non-Javadoc) | |
| 1599 | * | |
| 1600 | * @see jalview.datamodel.SequenceI#createDatasetSequence() | |
| 1601 | */ | |
| 1602 | 5905 | @Override |
| 1603 | public SequenceI createDatasetSequence() | |
| 1604 | { | |
| 1605 | 5905 | if (datasetSequence == null) |
| 1606 | { | |
| 1607 | 5904 | Sequence dsseq = new Sequence(getName(), |
| 1608 | AlignSeq.extractGaps(jalview.util.Comparison.GapChars, | |
| 1609 | getSequenceAsString()), | |
| 1610 | getStart(), getEnd()); | |
| 1611 | ||
| 1612 | 5904 | datasetSequence = dsseq; |
| 1613 | ||
| 1614 | 5904 | dsseq.setDescription(description); |
| 1615 | // move features and database references onto dataset sequence | |
| 1616 | 5904 | dsseq.sequenceFeatureStore = sequenceFeatureStore; |
| 1617 | 5904 | sequenceFeatureStore = null; |
| 1618 | 5904 | dsseq.dbrefs = dbrefs; |
| 1619 | 5904 | dbrefs = null; |
| 1620 | // TODO: search and replace any references to this sequence with | |
| 1621 | // references to the dataset sequence in Mappings on dbref | |
| 1622 | 5904 | dsseq.pdbIds = pdbIds; |
| 1623 | 5904 | pdbIds = null; |
| 1624 | 5904 | datasetSequence.updatePDBIds(); |
| 1625 | 5904 | if (annotation != null) |
| 1626 | { | |
| 1627 | // annotation is cloned rather than moved, to preserve what's currently | |
| 1628 | // on the alignment | |
| 1629 | 284 | for (AlignmentAnnotation aa : annotation) |
| 1630 | { | |
| 1631 | 328 | AlignmentAnnotation _aa = new AlignmentAnnotation(aa); |
| 1632 | 328 | _aa.sequenceRef = datasetSequence; |
| 1633 | 328 | _aa.adjustForAlignment(); // uses annotation's own record of |
| 1634 | // sequence-column mapping | |
| 1635 | 328 | datasetSequence.addAlignmentAnnotation(_aa); |
| 1636 | ||
| 1637 | 328 | if (_cmholder != null) |
| 1638 | { // transfer contact matrices | |
| 1639 | 1 | ContactMatrixI cm = _cmholder.getContactMatrixFor(aa); |
| 1640 | 1 | if (cm != null) |
| 1641 | { | |
| 1642 | 1 | datasetSequence.addContactListFor(_aa, cm); |
| 1643 | 1 | datasetSequence.addContactListFor(aa, cm); |
| 1644 | } | |
| 1645 | } | |
| 1646 | } | |
| 1647 | } | |
| 1648 | // all matrices should have been transferred. so we clear the local holder | |
| 1649 | 5904 | _cmholder = null; |
| 1650 | } | |
| 1651 | 5905 | return datasetSequence; |
| 1652 | } | |
| 1653 | ||
| 1654 | /* | |
| 1655 | * (non-Javadoc) | |
| 1656 | * | |
| 1657 | * @see | |
| 1658 | * jalview.datamodel.SequenceI#setAlignmentAnnotation(AlignmmentAnnotation[] | |
| 1659 | * annotations) | |
| 1660 | */ | |
| 1661 | 2 | @Override |
| 1662 | public void setAlignmentAnnotation(AlignmentAnnotation[] annotations) | |
| 1663 | { | |
| 1664 | 2 | if (annotation != null) |
| 1665 | { | |
| 1666 | 2 | annotation.removeAllElements(); |
| 1667 | } | |
| 1668 | 2 | if (annotations != null) |
| 1669 | { | |
| 1670 | 2 | for (int i = 0; i < annotations.length; i++) |
| 1671 | { | |
| 1672 | 0 | if (annotations[i] != null) |
| 1673 | { | |
| 1674 | 0 | addAlignmentAnnotation(annotations[i]); |
| 1675 | } | |
| 1676 | } | |
| 1677 | } | |
| 1678 | } | |
| 1679 | ||
| 1680 | 21565172 | @Override |
| 1681 | public AlignmentAnnotation[] getAnnotation(String label) | |
| 1682 | { | |
| 1683 | 21492638 | if (annotation == null || annotation.size() == 0) |
| 1684 | { | |
| 1685 | 21403210 | return null; |
| 1686 | } | |
| 1687 | ||
| 1688 | 200132 | Vector<AlignmentAnnotation> subset = new Vector<>(); |
| 1689 | 200132 | Enumeration<AlignmentAnnotation> e = annotation.elements(); |
| 1690 | 594812 | while (e.hasMoreElements()) |
| 1691 | { | |
| 1692 | 394680 | AlignmentAnnotation ann = e.nextElement(); |
| 1693 | 394680 | if (ann.label != null && ann.label.equals(label)) |
| 1694 | { | |
| 1695 | 95856 | subset.addElement(ann); |
| 1696 | } | |
| 1697 | } | |
| 1698 | 200132 | if (subset.size() == 0) |
| 1699 | { | |
| 1700 | 113045 | return null; |
| 1701 | } | |
| 1702 | 87087 | AlignmentAnnotation[] anns = new AlignmentAnnotation[subset.size()]; |
| 1703 | 87087 | int i = 0; |
| 1704 | 87087 | e = subset.elements(); |
| 1705 | 182943 | while (e.hasMoreElements()) |
| 1706 | { | |
| 1707 | 95856 | anns[i++] = e.nextElement(); |
| 1708 | } | |
| 1709 | 87087 | subset.removeAllElements(); |
| 1710 | 87087 | return anns; |
| 1711 | } | |
| 1712 | ||
| 1713 | 5949 | @Override |
| 1714 | public boolean updatePDBIds() | |
| 1715 | { | |
| 1716 | 5949 | if (datasetSequence != null) |
| 1717 | { | |
| 1718 | // TODO: could merge DBRefs | |
| 1719 | 22 | return datasetSequence.updatePDBIds(); |
| 1720 | } | |
| 1721 | 5927 | if (dbrefs == null || dbrefs.size() == 0) |
| 1722 | { | |
| 1723 | 5398 | return false; |
| 1724 | } | |
| 1725 | 529 | boolean added = false; |
| 1726 | 1940 | for (int ib = 0, nb = dbrefs.size(); ib < nb; ib++) |
| 1727 | { | |
| 1728 | 1411 | DBRefEntry dbr = dbrefs.get(ib); |
| 1729 | 1411 | if (DBRefSource.PDB.equals(dbr.getSource())) |
| 1730 | { | |
| 1731 | /* | |
| 1732 | * 'Add' any PDB dbrefs as a PDBEntry - add is only performed if the | |
| 1733 | * PDB id is not already present in a 'matching' PDBEntry | |
| 1734 | * Constructor parses out a chain code if appended to the accession id | |
| 1735 | * (a fudge used to 'store' the chain code in the DBRef) | |
| 1736 | */ | |
| 1737 | 208 | PDBEntry pdbe = new PDBEntry(dbr); |
| 1738 | 208 | added |= addPDBId(pdbe); |
| 1739 | } | |
| 1740 | } | |
| 1741 | 529 | return added; |
| 1742 | } | |
| 1743 | ||
| 1744 | 17 | @Override |
| 1745 | public void transferAnnotation(SequenceI entry, Mapping mp) | |
| 1746 | { | |
| 1747 | 17 | if (datasetSequence != null) |
| 1748 | { | |
| 1749 | 0 | datasetSequence.transferAnnotation(entry, mp); |
| 1750 | 0 | return; |
| 1751 | } | |
| 1752 | 17 | if (entry.getDatasetSequence() != null) |
| 1753 | { | |
| 1754 | 0 | transferAnnotation(entry.getDatasetSequence(), mp); |
| 1755 | 0 | return; |
| 1756 | } | |
| 1757 | // transfer from entry to sequence | |
| 1758 | // if entry has a description and sequence doesn't, then transfer | |
| 1759 | 17 | if (entry.getDescription() != null |
| 1760 | && (description == null || description.trim().length() == 0)) | |
| 1761 | { | |
| 1762 | 1 | description = entry.getDescription(); |
| 1763 | } | |
| 1764 | ||
| 1765 | // transfer any new features from entry onto sequence | |
| 1766 | 17 | if (entry.getSequenceFeatures() != null) |
| 1767 | { | |
| 1768 | ||
| 1769 | 17 | List<SequenceFeature> sfs = entry.getSequenceFeatures(); |
| 1770 | 17 | for (SequenceFeature feature : sfs) |
| 1771 | { | |
| 1772 | 3352 | SequenceFeature sf[] = (mp != null) ? mp.locateFeature(feature) |
| 1773 | : new SequenceFeature[] | |
| 1774 | { new SequenceFeature(feature) }; | |
| 1775 | 3352 | if (sf != null) |
| 1776 | { | |
| 1777 | 6704 | for (int sfi = 0; sfi < sf.length; sfi++) |
| 1778 | { | |
| 1779 | 3352 | addSequenceFeature(sf[sfi]); |
| 1780 | } | |
| 1781 | } | |
| 1782 | } | |
| 1783 | } | |
| 1784 | ||
| 1785 | // transfer PDB entries | |
| 1786 | 17 | if (entry.getAllPDBEntries() != null) |
| 1787 | { | |
| 1788 | 17 | Enumeration<PDBEntry> e = entry.getAllPDBEntries().elements(); |
| 1789 | 31 | while (e.hasMoreElements()) |
| 1790 | { | |
| 1791 | 14 | PDBEntry pdb = e.nextElement(); |
| 1792 | 14 | addPDBId(pdb); |
| 1793 | } | |
| 1794 | } | |
| 1795 | // transfer database references | |
| 1796 | 17 | List<DBRefEntry> entryRefs = entry.getDBRefs(); |
| 1797 | 17 | if (entryRefs != null) |
| 1798 | { | |
| 1799 | 34 | for (int r = 0, n = entryRefs.size(); r < n; r++) |
| 1800 | { | |
| 1801 | 17 | DBRefEntry newref = new DBRefEntry(entryRefs.get(r)); |
| 1802 | 17 | if (newref.getMap() != null && mp != null) |
| 1803 | { | |
| 1804 | // remap ref using our local mapping | |
| 1805 | } | |
| 1806 | // we also assume all version string setting is done by dbSourceProxy | |
| 1807 | /* | |
| 1808 | * if (!newref.getSource().equalsIgnoreCase(dbSource)) { | |
| 1809 | * newref.setSource(dbSource); } | |
| 1810 | */ | |
| 1811 | 17 | addDBRef(newref); |
| 1812 | } | |
| 1813 | } | |
| 1814 | } | |
| 1815 | ||
| 1816 | 3 | @Override |
| 1817 | public void setRNA(RNA r) | |
| 1818 | { | |
| 1819 | 3 | rna = r; |
| 1820 | } | |
| 1821 | ||
| 1822 | 0 | @Override |
| 1823 | public RNA getRNA() | |
| 1824 | { | |
| 1825 | 0 | return rna; |
| 1826 | } | |
| 1827 | ||
| 1828 | 6 | @Override |
| 1829 | public List<AlignmentAnnotation> getAlignmentAnnotations(String calcId, | |
| 1830 | String label) | |
| 1831 | { | |
| 1832 | 6 | return getAlignmentAnnotations(calcId, label, null, true); |
| 1833 | } | |
| 1834 | ||
| 1835 | 342 | @Override |
| 1836 | public List<AlignmentAnnotation> getAlignmentAnnotations(String calcId, | |
| 1837 | String label, String description) | |
| 1838 | { | |
| 1839 | 342 | return getAlignmentAnnotations(calcId, label, description, false); |
| 1840 | } | |
| 1841 | ||
| 1842 | 348 | private List<AlignmentAnnotation> getAlignmentAnnotations(String calcId, |
| 1843 | String label, String description, boolean ignoreDescription) | |
| 1844 | { | |
| 1845 | 348 | List<AlignmentAnnotation> result = new ArrayList<>(); |
| 1846 | 348 | if (this.annotation != null) |
| 1847 | { | |
| 1848 | 290 | for (AlignmentAnnotation ann : annotation) |
| 1849 | { | |
| 1850 | 758 | if ((ann.calcId != null && ann.calcId.equals(calcId)) |
| 1851 | && (ann.label != null && ann.label.equals(label)) | |
| 1852 | && ((ignoreDescription && description == null) | |
| 1853 | || (ann.description != null | |
| 1854 | && ann.description.equals(description)))) | |
| 1855 | ||
| 1856 | { | |
| 1857 | 161 | result.add(ann); |
| 1858 | } | |
| 1859 | } | |
| 1860 | } | |
| 1861 | 348 | return result; |
| 1862 | } | |
| 1863 | ||
| 1864 | 277 | @Override |
| 1865 | public String toString() | |
| 1866 | { | |
| 1867 | 277 | return getDisplayId(false); |
| 1868 | } | |
| 1869 | ||
| 1870 | 26 | @Override |
| 1871 | public PDBEntry getPDBEntry(String pdbIdStr) | |
| 1872 | { | |
| 1873 | 26 | if (getDatasetSequence() != null) |
| 1874 | { | |
| 1875 | 0 | return getDatasetSequence().getPDBEntry(pdbIdStr); |
| 1876 | } | |
| 1877 | 26 | if (pdbIds == null) |
| 1878 | { | |
| 1879 | 0 | return null; |
| 1880 | } | |
| 1881 | 26 | List<PDBEntry> entries = getAllPDBEntries(); |
| 1882 | 26 | for (PDBEntry entry : entries) |
| 1883 | { | |
| 1884 | 40 | if (entry.getId().equalsIgnoreCase(pdbIdStr)) |
| 1885 | { | |
| 1886 | 10 | return entry; |
| 1887 | } | |
| 1888 | } | |
| 1889 | 16 | return null; |
| 1890 | } | |
| 1891 | ||
| 1892 | private List<DBRefEntry> tmpList; | |
| 1893 | ||
| 1894 | 578 | @Override |
| 1895 | public List<DBRefEntry> getPrimaryDBRefs() | |
| 1896 | { | |
| 1897 | 578 | if (datasetSequence != null) |
| 1898 | { | |
| 1899 | 276 | return datasetSequence.getPrimaryDBRefs(); |
| 1900 | } | |
| 1901 | 302 | if (dbrefs == null || dbrefs.size() == 0) |
| 1902 | { | |
| 1903 | 9 | return Collections.emptyList(); |
| 1904 | } | |
| 1905 | 293 | synchronized (dbrefs) |
| 1906 | { | |
| 1907 | 293 | if (refModCount == dbrefs.getModCount() && primaryRefs != null) |
| 1908 | { | |
| 1909 | 5 | return primaryRefs; // no changes |
| 1910 | } | |
| 1911 | 288 | refModCount = dbrefs.getModCount(); |
| 1912 | 288 | List<DBRefEntry> primaries = (primaryRefs == null |
| 1913 | ? (primaryRefs = new ArrayList<>()) | |
| 1914 | : primaryRefs); | |
| 1915 | 288 | primaries.clear(); |
| 1916 | 288 | if (tmpList == null) |
| 1917 | { | |
| 1918 | 286 | tmpList = new ArrayList<>(); |
| 1919 | 286 | tmpList.add(null); // for replacement |
| 1920 | } | |
| 1921 | 1147 | for (int i = 0, n = dbrefs.size(); i < n; i++) |
| 1922 | { | |
| 1923 | 859 | DBRefEntry ref = dbrefs.get(i); |
| 1924 | 859 | if (!ref.isPrimaryCandidate()) |
| 1925 | { | |
| 1926 | 807 | continue; |
| 1927 | } | |
| 1928 | 52 | if (ref.hasMap()) |
| 1929 | { | |
| 1930 | 5 | MapList mp = ref.getMap().getMap(); |
| 1931 | 5 | if (mp.getFromLowest() > start || mp.getFromHighest() < end) |
| 1932 | { | |
| 1933 | // map only involves a subsequence, so cannot be primary | |
| 1934 | 2 | continue; |
| 1935 | } | |
| 1936 | } | |
| 1937 | // whilst it looks like it is a primary ref, we also sanity check type | |
| 1938 | 50 | if (DBRefSource.PDB_CANONICAL_NAME |
| 1939 | .equals(ref.getCanonicalSourceName())) | |
| 1940 | { | |
| 1941 | // PDB dbrefs imply there should be a PDBEntry associated | |
| 1942 | // TODO: tighten PDB dbrefs | |
| 1943 | // formally imply Jalview has actually downloaded and | |
| 1944 | // parsed the pdb file. That means there should be a cached file | |
| 1945 | // handle on the PDBEntry, and a real mapping between sequence and | |
| 1946 | // extracted sequence from PDB file | |
| 1947 | 21 | PDBEntry pdbentry = getPDBEntry(ref.getAccessionId()); |
| 1948 | 21 | if (pdbentry == null || pdbentry.getFile() == null) |
| 1949 | { | |
| 1950 | 18 | continue; |
| 1951 | } | |
| 1952 | } | |
| 1953 | else | |
| 1954 | { | |
| 1955 | // check standard protein or dna sources | |
| 1956 | 29 | tmpList.set(0, ref); |
| 1957 | 29 | List<DBRefEntry> res = DBRefUtils.selectDbRefs(!isProtein(), |
| 1958 | tmpList); | |
| 1959 | 29 | if (res == null || res.get(0) != tmpList.get(0)) |
| 1960 | { | |
| 1961 | 6 | continue; |
| 1962 | } | |
| 1963 | } | |
| 1964 | 26 | primaries.add(ref); |
| 1965 | } | |
| 1966 | ||
| 1967 | // version must be not null, as otherwise it will not be a candidate, | |
| 1968 | // above | |
| 1969 | 288 | DBRefUtils.ensurePrimaries(this, primaries); |
| 1970 | 288 | return primaries; |
| 1971 | } | |
| 1972 | } | |
| 1973 | ||
| 1974 | /** | |
| 1975 | * {@inheritDoc} | |
| 1976 | */ | |
| 1977 | 260823 | @Override |
| 1978 | public List<SequenceFeature> findFeatures(int fromColumn, int toColumn, | |
| 1979 | String... types) | |
| 1980 | { | |
| 1981 | 260834 | int startPos = findPosition(fromColumn - 1); // convert base 1 to base 0 |
| 1982 | 260816 | int endPos = fromColumn == toColumn ? startPos |
| 1983 | : findPosition(toColumn - 1); | |
| 1984 | ||
| 1985 | 260794 | List<SequenceFeature> result = getFeatures().findFeatures(startPos, |
| 1986 | endPos, types); | |
| 1987 | ||
| 1988 | /* | |
| 1989 | * if end column is gapped, endPos may be to the right, | |
| 1990 | * and we may have included adjacent or enclosing features; | |
| 1991 | * remove any that are not enclosing, non-contact features | |
| 1992 | */ | |
| 1993 | 260600 | boolean endColumnIsGapped = toColumn > 0 && toColumn <= sequence.length |
| 1994 | && Comparison.isGap(sequence[toColumn - 1]); | |
| 1995 | 260626 | if (endPos > this.end || endColumnIsGapped) |
| 1996 | { | |
| 1997 | 65 | ListIterator<SequenceFeature> it = result.listIterator(); |
| 1998 | 474 | while (it.hasNext()) |
| 1999 | { | |
| 2000 | 409 | SequenceFeature sf = it.next(); |
| 2001 | 409 | int sfBegin = sf.getBegin(); |
| 2002 | 409 | int sfEnd = sf.getEnd(); |
| 2003 | 409 | int featureStartColumn = findIndex(sfBegin); |
| 2004 | 409 | if (featureStartColumn > toColumn) |
| 2005 | { | |
| 2006 | 3 | it.remove(); |
| 2007 | } | |
| 2008 | 406 | else if (featureStartColumn < fromColumn) |
| 2009 | { | |
| 2010 | 7 | int featureEndColumn = sfEnd == sfBegin ? featureStartColumn |
| 2011 | : findIndex(sfEnd); | |
| 2012 | 7 | if (featureEndColumn < fromColumn) |
| 2013 | { | |
| 2014 | 0 | it.remove(); |
| 2015 | } | |
| 2016 | 7 | else if (featureEndColumn > toColumn && sf.isContactFeature()) |
| 2017 | { | |
| 2018 | /* | |
| 2019 | * remove an enclosing feature if it is a contact feature | |
| 2020 | */ | |
| 2021 | 3 | it.remove(); |
| 2022 | } | |
| 2023 | } | |
| 2024 | } | |
| 2025 | } | |
| 2026 | ||
| 2027 | 260601 | return result; |
| 2028 | } | |
| 2029 | ||
| 2030 | /** | |
| 2031 | * Invalidates any stale cursors (forcing recalculation) by incrementing the | |
| 2032 | * token that has to match the one presented by the cursor | |
| 2033 | */ | |
| 2034 | 14348 | @Override |
| 2035 | public void sequenceChanged() | |
| 2036 | { | |
| 2037 | 14348 | changeCount++; |
| 2038 | } | |
| 2039 | ||
| 2040 | /** | |
| 2041 | * {@inheritDoc} | |
| 2042 | */ | |
| 2043 | 6 | @Override |
| 2044 | public int replace(char c1, char c2) | |
| 2045 | { | |
| 2046 | 6 | if (c1 == c2) |
| 2047 | { | |
| 2048 | 1 | return 0; |
| 2049 | } | |
| 2050 | 5 | int count = 0; |
| 2051 | 5 | synchronized (sequence) |
| 2052 | { | |
| 2053 | 57 | for (int c = 0; c < sequence.length; c++) |
| 2054 | { | |
| 2055 | 52 | if (sequence[c] == c1) |
| 2056 | { | |
| 2057 | 12 | sequence[c] = c2; |
| 2058 | 12 | count++; |
| 2059 | } | |
| 2060 | } | |
| 2061 | } | |
| 2062 | 5 | if (count > 0) |
| 2063 | { | |
| 2064 | 4 | sequenceChanged(); |
| 2065 | } | |
| 2066 | ||
| 2067 | 5 | return count; |
| 2068 | } | |
| 2069 | ||
| 2070 | 189 | @Override |
| 2071 | public String getSequenceStringFromIterator(Iterator<int[]> it) | |
| 2072 | { | |
| 2073 | 189 | StringBuilder newSequence = new StringBuilder(); |
| 2074 | 382 | while (it.hasNext()) |
| 2075 | { | |
| 2076 | 193 | int[] block = it.next(); |
| 2077 | 193 | if (it.hasNext()) |
| 2078 | { | |
| 2079 | 4 | newSequence.append(getSequence(block[0], block[1] + 1)); |
| 2080 | } | |
| 2081 | else | |
| 2082 | { | |
| 2083 | 189 | newSequence.append(getSequence(block[0], block[1])); |
| 2084 | } | |
| 2085 | } | |
| 2086 | ||
| 2087 | 189 | return newSequence.toString(); |
| 2088 | } | |
| 2089 | ||
| 2090 | 170 | @Override |
| 2091 | public int firstResidueOutsideIterator(Iterator<int[]> regions) | |
| 2092 | { | |
| 2093 | 170 | int start = 0; |
| 2094 | ||
| 2095 | 170 | if (!regions.hasNext()) |
| 2096 | { | |
| 2097 | 123 | return findIndex(getStart()) - 1; |
| 2098 | } | |
| 2099 | ||
| 2100 | // Simply walk along the sequence whilst watching for region | |
| 2101 | // boundaries | |
| 2102 | 47 | int hideStart = getLength(); |
| 2103 | 47 | int hideEnd = -1; |
| 2104 | 47 | boolean foundStart = false; |
| 2105 | ||
| 2106 | // step through the non-gapped positions of the sequence | |
| 2107 | 1227 | for (int i = getStart(); i <= getEnd() && (!foundStart); i++) |
| 2108 | { | |
| 2109 | // get alignment position of this residue in the sequence | |
| 2110 | 1180 | int p = findIndex(i) - 1; |
| 2111 | ||
| 2112 | // update region start/end | |
| 2113 | 1260 | while (hideEnd < p && regions.hasNext()) |
| 2114 | { | |
| 2115 | 80 | int[] region = regions.next(); |
| 2116 | 80 | hideStart = region[0]; |
| 2117 | 80 | hideEnd = region[1]; |
| 2118 | } | |
| 2119 | 1180 | if (hideEnd < p) |
| 2120 | { | |
| 2121 | 7 | hideStart = getLength(); |
| 2122 | } | |
| 2123 | // update boundary for sequence | |
| 2124 | 1180 | if (p < hideStart) |
| 2125 | { | |
| 2126 | 35 | start = p; |
| 2127 | 35 | foundStart = true; |
| 2128 | } | |
| 2129 | } | |
| 2130 | ||
| 2131 | 47 | if (foundStart) |
| 2132 | { | |
| 2133 | 35 | return start; |
| 2134 | } | |
| 2135 | // otherwise, sequence was completely hidden | |
| 2136 | 12 | return 0; |
| 2137 | } | |
| 2138 | ||
| 2139 | //// | |
| 2140 | //// Contact Matrix Holder Boilerplate | |
| 2141 | //// | |
| 2142 | ContactMapHolderI _cmholder = null; | |
| 2143 | ||
| 2144 | 8083 | private ContactMapHolderI getContactMapHolder() |
| 2145 | { | |
| 2146 | 8083 | if (datasetSequence != null) |
| 2147 | { | |
| 2148 | 3755 | return ((Sequence) datasetSequence).getContactMapHolder(); |
| 2149 | } | |
| 2150 | 4328 | if (_cmholder == null) |
| 2151 | { | |
| 2152 | 190 | _cmholder = new ContactMapHolder(); |
| 2153 | } | |
| 2154 | 4328 | return _cmholder; |
| 2155 | } | |
| 2156 | ||
| 2157 | 12 | @Override |
| 2158 | public Collection<ContactMatrixI> getContactMaps() | |
| 2159 | { | |
| 2160 | 12 | return getContactMapHolder().getContactMaps(); |
| 2161 | } | |
| 2162 | ||
| 2163 | 444 | @Override |
| 2164 | public ContactMatrixI getContactMatrixFor(AlignmentAnnotation ann) | |
| 2165 | { | |
| 2166 | 444 | return getContactMapHolder().getContactMatrixFor(ann); |
| 2167 | } | |
| 2168 | ||
| 2169 | 3680 | @Override |
| 2170 | public ContactListI getContactListFor(AlignmentAnnotation _aa, int column) | |
| 2171 | { | |
| 2172 | 3680 | return getContactMapHolder().getContactListFor(_aa, column); |
| 2173 | } | |
| 2174 | ||
| 2175 | 120 | @Override |
| 2176 | public AlignmentAnnotation addContactList(ContactMatrixI cm) | |
| 2177 | { | |
| 2178 | 120 | AlignmentAnnotation aa; |
| 2179 | ||
| 2180 | 120 | if (datasetSequence != null) |
| 2181 | { | |
| 2182 | 2 | aa = datasetSequence.addContactList(cm); |
| 2183 | // clone the annotation for the local sequence | |
| 2184 | 2 | aa = new AlignmentAnnotation(aa); |
| 2185 | 2 | aa.restrict(start, end); |
| 2186 | 2 | aa.adjustForAlignment(); |
| 2187 | 2 | getContactMapHolder().addContactListFor(aa, cm); |
| 2188 | 2 | addAlignmentAnnotation(aa); |
| 2189 | 2 | return aa; |
| 2190 | } | |
| 2191 | ||
| 2192 | // construct new annotation for matrix on dataset sequence | |
| 2193 | 118 | aa = getContactMapHolder().addContactList(cm); |
| 2194 | ||
| 2195 | 118 | Annotation _aa[] = new Annotation[getLength()]; |
| 2196 | ||
| 2197 | 61805 | for (int i = 0; i < _aa.length; _aa[i++] = new Annotation(0.0f)) |
| 2198 | { | |
| 2199 | 61687 | ; |
| 2200 | } | |
| 2201 | 118 | aa.annotations = _aa; |
| 2202 | 118 | aa.setSequenceRef(this); |
| 2203 | 118 | if (cm instanceof MappableContactMatrix |
| 2204 | && !((MappableContactMatrix) cm).hasReferenceSeq()) | |
| 2205 | { | |
| 2206 | 63 | ((MappableContactMatrix) cm).setRefSeq(this); |
| 2207 | } | |
| 2208 | 118 | aa.createSequenceMapping(this, getStart(), false); |
| 2209 | 118 | addAlignmentAnnotation(aa); |
| 2210 | 118 | return aa; |
| 2211 | } | |
| 2212 | ||
| 2213 | 72 | @Override |
| 2214 | public void addContactListFor(AlignmentAnnotation annotation, | |
| 2215 | ContactMatrixI cm) | |
| 2216 | { | |
| 2217 | 72 | getContactMapHolder().addContactListFor(annotation, cm); |
| 2218 | } | |
| 2219 | } |