jalviewX
File JmolParser.java
Coverage histogram
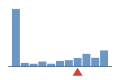
28% of files have more coverage
Classes
| Class | Line # | Actions | ||||
|---|---|---|---|---|---|---|
| JmolParser | 58 | 213 | 97 | 116 |
Contributing tests
This file is covered by 18 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | package jalview.ext.jmol; | |
| 22 | ||
| 23 | import jalview.datamodel.AlignmentAnnotation; | |
| 24 | import jalview.datamodel.Annotation; | |
| 25 | import jalview.datamodel.PDBEntry; | |
| 26 | import jalview.datamodel.SequenceI; | |
| 27 | import jalview.io.DataSourceType; | |
| 28 | import jalview.io.FileParse; | |
| 29 | import jalview.io.StructureFile; | |
| 30 | import jalview.schemes.ResidueProperties; | |
| 31 | import jalview.util.Format; | |
| 32 | import jalview.util.MessageManager; | |
| 33 | ||
| 34 | import java.io.IOException; | |
| 35 | import java.util.ArrayList; | |
| 36 | import java.util.HashMap; | |
| 37 | import java.util.List; | |
| 38 | import java.util.Map; | |
| 39 | import java.util.Vector; | |
| 40 | ||
| 41 | import org.jmol.api.JmolStatusListener; | |
| 42 | import org.jmol.api.JmolViewer; | |
| 43 | import org.jmol.c.CBK; | |
| 44 | import org.jmol.c.STR; | |
| 45 | import org.jmol.modelset.ModelSet; | |
| 46 | import org.jmol.viewer.Viewer; | |
| 47 | ||
| 48 | import mc_view.Atom; | |
| 49 | import mc_view.PDBChain; | |
| 50 | import mc_view.Residue; | |
| 51 | ||
| 52 | /** | |
| 53 | * Import and process files with Jmol for file like PDB, mmCIF | |
| 54 | * | |
| 55 | * @author jprocter | |
| 56 | * | |
| 57 | */ | |
| 58 | public class JmolParser extends StructureFile implements JmolStatusListener | |
| 59 | { | |
| 60 | Viewer viewer = null; | |
| 61 | ||
| 62 | 76 |  public JmolParser(boolean immediate, Object inFile, public JmolParser(boolean immediate, Object inFile, |
| 63 | DataSourceType sourceType) throws IOException | |
| 64 | { | |
| 65 | // BH 2018 File or String for filename | |
| 66 | 76 | super(immediate, inFile, sourceType); |
| 67 | } | |
| 68 | ||
| 69 | 14 | public JmolParser(Object inFile, DataSourceType sourceType) |
| 70 | throws IOException | |
| 71 | { | |
| 72 | 14 | super(inFile, sourceType); |
| 73 | } | |
| 74 | ||
| 75 | 2 | public JmolParser(FileParse fp) throws IOException |
| 76 | { | |
| 77 | 2 | super(fp); |
| 78 | } | |
| 79 | ||
| 80 | 1 | public JmolParser() |
| 81 | { | |
| 82 | } | |
| 83 | ||
| 84 | /** | |
| 85 | * Calls the Jmol library to parse the PDB/mmCIF file, and then inspects the | |
| 86 | * resulting object model to generate Jalview-style sequences, with secondary | |
| 87 | * structure annotation added where available (i.e. where it has been computed | |
| 88 | * by Jmol using DSSP). | |
| 89 | * | |
| 90 | * @see jalview.io.AlignFile#parse() | |
| 91 | */ | |
| 92 | 92 | @Override |
| 93 | public void parse() throws IOException | |
| 94 | { | |
| 95 | 92 | setChains(new Vector<PDBChain>()); |
| 96 | 92 | Viewer jmolModel = getJmolData(); |
| 97 | 92 | jmolModel.openReader(getDataName(), getDataName(), getReader()); |
| 98 | 92 | waitForScript(jmolModel); |
| 99 | ||
| 100 | /* | |
| 101 | * Convert one or more Jmol Model objects to Jalview sequences | |
| 102 | */ | |
| 103 | 92 | if (jmolModel.ms.mc > 0) |
| 104 | { | |
| 105 | // ideally we do this | |
| 106 | // try | |
| 107 | // { | |
| 108 | // setStructureFileType(jmolModel.evalString("show _fileType")); | |
| 109 | // } catch (Exception q) | |
| 110 | // { | |
| 111 | // } | |
| 112 | // ; | |
| 113 | // instead, we distinguish .cif from non-.cif by filename | |
| 114 | 92 | setStructureFileType(getDataName().toLowerCase().endsWith(".cif") |
| 115 | ? PDBEntry.Type.MMCIF.toString() | |
| 116 | : "PDB"); | |
| 117 | ||
| 118 | 92 | transformJmolModelToJalview(jmolModel.ms); |
| 119 | } | |
| 120 | } | |
| 121 | ||
| 122 | /** | |
| 123 | * create a headless jmol instance for dataprocessing | |
| 124 | * | |
| 125 | * @return | |
| 126 | */ | |
| 127 | 92 | private Viewer getJmolData() |
| 128 | { | |
| 129 | 92 | if (viewer == null) |
| 130 | { | |
| 131 | 92 | try |
| 132 | { | |
| 133 | /* | |
| 134 | * params -o (output to sysout) -n (nodisplay) -x (exit when finished) | |
| 135 | * see http://wiki.jmol.org/index.php/Jmol_Application | |
| 136 | */ | |
| 137 | ||
| 138 | 92 | viewer = JalviewJmolBinding.getJmolData(this); |
| 139 | // ensure the 'new' (DSSP) not 'old' (Ramachandran) SS method is used | |
| 140 | 92 | viewer.setBooleanProperty("defaultStructureDSSP", true); |
| 141 | } catch (ClassCastException x) | |
| 142 | { | |
| 143 | 0 | throw new Error(MessageManager.formatMessage( |
| 144 | "error.jmol_version_not_compatible_with_jalview_version", | |
| 145 | new String[] | |
| 146 | { JmolViewer.getJmolVersion() }), x); | |
| 147 | } | |
| 148 | } | |
| 149 | 92 | return viewer; |
| 150 | } | |
| 151 | ||
| 152 | 92 | public void transformJmolModelToJalview(ModelSet ms) throws IOException |
| 153 | { | |
| 154 | 92 | try |
| 155 | { | |
| 156 | 92 | String lastID = ""; |
| 157 | 92 | List<SequenceI> rna = new ArrayList<SequenceI>(); |
| 158 | 92 | List<SequenceI> prot = new ArrayList<SequenceI>(); |
| 159 | 92 | PDBChain tmpchain; |
| 160 | 92 | String pdbId = (String) ms.getInfo(0, "title"); |
| 161 | ||
| 162 | 92 | if (pdbId == null) |
| 163 | { | |
| 164 | 1 | setId(safeName(getDataName())); |
| 165 | 1 | setPDBIdAvailable(false); |
| 166 | } | |
| 167 | else | |
| 168 | { | |
| 169 | 91 | setId(pdbId); |
| 170 | 91 | setPDBIdAvailable(true); |
| 171 | } | |
| 172 | 92 | List<Atom> significantAtoms = convertSignificantAtoms(ms); |
| 173 | 92 | for (Atom tmpatom : significantAtoms) |
| 174 | { | |
| 175 | 19112 | try |
| 176 | { | |
| 177 | 19112 | tmpchain = findChain(tmpatom.chain); |
| 178 | 18975 | if (tmpatom.resNumIns.trim().equals(lastID)) |
| 179 | { | |
| 180 | // phosphorylated protein - seen both CA and P.. | |
| 181 | 1 | continue; |
| 182 | } | |
| 183 | 18974 | tmpchain.atoms.addElement(tmpatom); |
| 184 | } catch (Exception e) | |
| 185 | { | |
| 186 | 137 | tmpchain = new PDBChain(getId(), tmpatom.chain); |
| 187 | 137 | getChains().add(tmpchain); |
| 188 | 137 | tmpchain.atoms.addElement(tmpatom); |
| 189 | } | |
| 190 | 19111 | lastID = tmpatom.resNumIns.trim(); |
| 191 | } | |
| 192 | 92 | if (isParseImmediately()) |
| 193 | { | |
| 194 | // configure parsing settings from the static singleton | |
| 195 | 92 | xferSettings(); |
| 196 | } | |
| 197 | ||
| 198 | 92 | makeResidueList(); |
| 199 | 92 | makeCaBondList(); |
| 200 | ||
| 201 | 92 | for (PDBChain chain : getChains()) |
| 202 | { | |
| 203 | 137 | SequenceI chainseq = postProcessChain(chain); |
| 204 | 137 | if (isRNA(chainseq)) |
| 205 | { | |
| 206 | 0 | rna.add(chainseq); |
| 207 | } | |
| 208 | else | |
| 209 | { | |
| 210 | 137 | prot.add(chainseq); |
| 211 | } | |
| 212 | ||
| 213 | // look at local setting for adding secondary tructure | |
| 214 | 137 | if (predictSecondaryStructure) |
| 215 | { | |
| 216 | 125 | createAnnotation(chainseq, chain, ms.at); |
| 217 | } | |
| 218 | } | |
| 219 | } catch (OutOfMemoryError er) | |
| 220 | { | |
| 221 | 0 | System.out.println( |
| 222 | "OUT OF MEMORY LOADING TRANSFORMING JMOL MODEL TO JALVIEW MODEL"); | |
| 223 | 0 | throw new IOException(MessageManager |
| 224 | .getString("exception.outofmemory_loading_mmcif_file")); | |
| 225 | } | |
| 226 | } | |
| 227 | ||
| 228 | 92 | private List<Atom> convertSignificantAtoms(ModelSet ms) |
| 229 | { | |
| 230 | 92 | List<Atom> significantAtoms = new ArrayList<Atom>(); |
| 231 | 92 | HashMap<String, org.jmol.modelset.Atom> chainTerMap = new HashMap<String, org.jmol.modelset.Atom>(); |
| 232 | 92 | org.jmol.modelset.Atom prevAtom = null; |
| 233 | 92 | for (org.jmol.modelset.Atom atom : ms.at) |
| 234 | { | |
| 235 | 102399 | if (atom.getAtomName().equalsIgnoreCase("CA") |
| 236 | || atom.getAtomName().equalsIgnoreCase("P")) | |
| 237 | { | |
| 238 | 19126 | if (!atomValidated(atom, prevAtom, chainTerMap)) |
| 239 | { | |
| 240 | 12 | continue; |
| 241 | } | |
| 242 | 19114 | Atom curAtom = new Atom(atom.x, atom.y, atom.z); |
| 243 | 19114 | curAtom.atomIndex = atom.getIndex(); |
| 244 | 19114 | curAtom.chain = atom.getChainIDStr(); |
| 245 | 19114 | curAtom.insCode = atom.group.getInsertionCode() == '\000' ? ' ' |
| 246 | : atom.group.getInsertionCode(); | |
| 247 | 19114 | curAtom.name = atom.getAtomName(); |
| 248 | 19114 | curAtom.number = atom.getAtomNumber(); |
| 249 | 19114 | curAtom.resName = atom.getGroup3(true); |
| 250 | 19114 | curAtom.resNumber = atom.getResno(); |
| 251 | 19114 | curAtom.occupancy = ms.occupancies != null |
| 252 | ? ms.occupancies[atom.getIndex()] | |
| 253 | : Float.valueOf(atom.getOccupancy100()); | |
| 254 | 19114 | String fmt = new Format("%4i").form(curAtom.resNumber); |
| 255 | 19114 | curAtom.resNumIns = (fmt + curAtom.insCode); |
| 256 | 19114 | curAtom.tfactor = atom.getBfactor100() / 100f; |
| 257 | 19114 | curAtom.type = 0; |
| 258 | // significantAtoms.add(curAtom); | |
| 259 | // ignore atoms from subsequent models | |
| 260 | 19114 | if (!significantAtoms.contains(curAtom)) |
| 261 | { | |
| 262 | 19112 | significantAtoms.add(curAtom); |
| 263 | } | |
| 264 | 19114 | prevAtom = atom; |
| 265 | } | |
| 266 | } | |
| 267 | 92 | return significantAtoms; |
| 268 | } | |
| 269 | ||
| 270 | 19126 | private boolean atomValidated(org.jmol.modelset.Atom curAtom, |
| 271 | org.jmol.modelset.Atom prevAtom, | |
| 272 | HashMap<String, org.jmol.modelset.Atom> chainTerMap) | |
| 273 | { | |
| 274 | // System.out.println("Atom: " + curAtom.getAtomNumber() | |
| 275 | // + " Last atom index " + curAtom.group.lastAtomIndex); | |
| 276 | 19126 | if (chainTerMap == null || prevAtom == null) |
| 277 | { | |
| 278 | 92 | return true; |
| 279 | } | |
| 280 | 19034 | String curAtomChId = curAtom.getChainIDStr(); |
| 281 | 19034 | String prevAtomChId = prevAtom.getChainIDStr(); |
| 282 | // new chain encoutered | |
| 283 | 19034 | if (!prevAtomChId.equals(curAtomChId)) |
| 284 | { | |
| 285 | // On chain switch add previous chain termination to xTerMap if not exists | |
| 286 | 57 | if (!chainTerMap.containsKey(prevAtomChId)) |
| 287 | { | |
| 288 | 51 | chainTerMap.put(prevAtomChId, prevAtom); |
| 289 | } | |
| 290 | // if current atom belongs to an already terminated chain and the resNum | |
| 291 | // diff < 5 then mark as valid and update termination Atom | |
| 292 | 57 | if (chainTerMap.containsKey(curAtomChId)) |
| 293 | { | |
| 294 | 12 | if (curAtom.getResno() < chainTerMap.get(curAtomChId).getResno()) |
| 295 | { | |
| 296 | 0 | return false; |
| 297 | } | |
| 298 | 12 | if ((curAtom.getResno() |
| 299 | - chainTerMap.get(curAtomChId).getResno()) < 5) | |
| 300 | { | |
| 301 | 0 | chainTerMap.put(curAtomChId, curAtom); |
| 302 | 0 | return true; |
| 303 | } | |
| 304 | 12 | return false; |
| 305 | } | |
| 306 | } | |
| 307 | // atom with previously terminated chain encountered | |
| 308 | 18977 | else if (chainTerMap.containsKey(curAtomChId)) |
| 309 | { | |
| 310 | 0 | if (curAtom.getResno() < chainTerMap.get(curAtomChId).getResno()) |
| 311 | { | |
| 312 | 0 | return false; |
| 313 | } | |
| 314 | 0 | if ((curAtom.getResno() |
| 315 | - chainTerMap.get(curAtomChId).getResno()) < 5) | |
| 316 | { | |
| 317 | 0 | chainTerMap.put(curAtomChId, curAtom); |
| 318 | 0 | return true; |
| 319 | } | |
| 320 | 0 | return false; |
| 321 | } | |
| 322 | // HETATM with resNum jump > 2 | |
| 323 | 19022 | return !(curAtom.isHetero() |
| 324 | && ((curAtom.getResno() - prevAtom.getResno()) > 2)); | |
| 325 | } | |
| 326 | ||
| 327 | 125 | private void createAnnotation(SequenceI sequence, PDBChain chain, |
| 328 | org.jmol.modelset.Atom[] jmolAtoms) | |
| 329 | { | |
| 330 | 125 | char[] secstr = new char[sequence.getLength()]; |
| 331 | 125 | char[] secstrcode = new char[sequence.getLength()]; |
| 332 | ||
| 333 | // Ensure Residue size equals Seq size | |
| 334 | 125 | if (chain.residues.size() != sequence.getLength()) |
| 335 | { | |
| 336 | 0 | return; |
| 337 | } | |
| 338 | 125 | int annotIndex = 0; |
| 339 | 125 | for (Residue residue : chain.residues) |
| 340 | { | |
| 341 | 19051 | Atom repAtom = residue.getAtoms().get(0); |
| 342 | 19051 | STR proteinStructureSubType = jmolAtoms[repAtom.atomIndex].group |
| 343 | .getProteinStructureSubType(); | |
| 344 | 19051 | setSecondaryStructure(proteinStructureSubType, annotIndex, secstr, |
| 345 | secstrcode); | |
| 346 | 19051 | ++annotIndex; |
| 347 | } | |
| 348 | 125 | addSecondaryStructureAnnotation(chain.pdbid, sequence, secstr, |
| 349 | secstrcode, chain.id, sequence.getStart()); | |
| 350 | } | |
| 351 | ||
| 352 | /** | |
| 353 | * Helper method that adds an AlignmentAnnotation for secondary structure to | |
| 354 | * the sequence, provided at least one secondary structure prediction has been | |
| 355 | * made | |
| 356 | * | |
| 357 | * @param modelTitle | |
| 358 | * @param seq | |
| 359 | * @param secstr | |
| 360 | * @param secstrcode | |
| 361 | * @param chainId | |
| 362 | * @param firstResNum | |
| 363 | * @return | |
| 364 | */ | |
| 365 | 125 | protected void addSecondaryStructureAnnotation(String modelTitle, |
| 366 | SequenceI sq, char[] secstr, char[] secstrcode, String chainId, | |
| 367 | int firstResNum) | |
| 368 | { | |
| 369 | 125 | int length = sq.getLength(); |
| 370 | 125 | boolean ssFound = false; |
| 371 | 125 | Annotation asecstr[] = new Annotation[length + firstResNum - 1]; |
| 372 | 19176 | for (int p = 0; p < length; p++) |
| 373 | { | |
| 374 | 19051 | if (secstr[p] >= 'A' && secstr[p] <= 'z') |
| 375 | { | |
| 376 | 7446 | try |
| 377 | { | |
| 378 | 7446 | asecstr[p] = new Annotation(String.valueOf(secstr[p]), null, |
| 379 | secstrcode[p], Float.NaN); | |
| 380 | 7446 | ssFound = true; |
| 381 | } catch (Exception e) | |
| 382 | { | |
| 383 | // e.printStackTrace(); | |
| 384 | } | |
| 385 | } | |
| 386 | } | |
| 387 | ||
| 388 | 125 | if (ssFound) |
| 389 | { | |
| 390 | 119 | String mt = modelTitle == null ? getDataName() : modelTitle; |
| 391 | 119 | mt += chainId; |
| 392 | 119 | AlignmentAnnotation ann = new AlignmentAnnotation( |
| 393 | "Secondary Structure", "Secondary Structure for " + mt, | |
| 394 | asecstr); | |
| 395 | 119 | ann.belowAlignment = true; |
| 396 | 119 | ann.visible = true; |
| 397 | 119 | ann.autoCalculated = false; |
| 398 | 119 | ann.setCalcId(getClass().getName()); |
| 399 | 119 | ann.adjustForAlignment(); |
| 400 | 119 | ann.validateRangeAndDisplay(); |
| 401 | 119 | annotations.add(ann); |
| 402 | 119 | sq.addAlignmentAnnotation(ann); |
| 403 | } | |
| 404 | } | |
| 405 | ||
| 406 | 92 | private void waitForScript(Viewer jmd) |
| 407 | { | |
| 408 | 92 | while (jmd.isScriptExecuting()) |
| 409 | { | |
| 410 | 0 | try |
| 411 | { | |
| 412 | 0 | Thread.sleep(50); |
| 413 | ||
| 414 | } catch (InterruptedException x) | |
| 415 | { | |
| 416 | } | |
| 417 | } | |
| 418 | } | |
| 419 | ||
| 420 | /** | |
| 421 | * Convert Jmol's secondary structure code to Jalview's, and stored it in the | |
| 422 | * secondary structure arrays at the given sequence position | |
| 423 | * | |
| 424 | * @param proteinStructureSubType | |
| 425 | * @param pos | |
| 426 | * @param secstr | |
| 427 | * @param secstrcode | |
| 428 | */ | |
| 429 | 19057 | protected void setSecondaryStructure(STR proteinStructureSubType, int pos, |
| 430 | char[] secstr, char[] secstrcode) | |
| 431 | { | |
| 432 | 19057 | switch (proteinStructureSubType) |
| 433 | { | |
| 434 | 449 | case HELIX310: |
| 435 | 449 | secstr[pos] = '3'; |
| 436 | 449 | break; |
| 437 | 1915 | case HELIX: |
| 438 | 2247 | case HELIXALPHA: |
| 439 | 4162 | secstr[pos] = 'H'; |
| 440 | 4162 | break; |
| 441 | 1 | case HELIXPI: |
| 442 | 1 | secstr[pos] = 'P'; |
| 443 | 1 | break; |
| 444 | 3287 | case SHEET: |
| 445 | 3287 | secstr[pos] = 'E'; |
| 446 | 3287 | break; |
| 447 | 11158 | default: |
| 448 | 11158 | secstr[pos] = 0; |
| 449 | } | |
| 450 | ||
| 451 | 19057 | switch (proteinStructureSubType) |
| 452 | { | |
| 453 | 449 | case HELIX310: |
| 454 | 2247 | case HELIXALPHA: |
| 455 | 1 | case HELIXPI: |
| 456 | 1915 | case HELIX: |
| 457 | 4612 | secstrcode[pos] = 'H'; |
| 458 | 4612 | break; |
| 459 | 3287 | case SHEET: |
| 460 | 3287 | secstrcode[pos] = 'E'; |
| 461 | 3287 | break; |
| 462 | 11158 | default: |
| 463 | 11158 | secstrcode[pos] = 0; |
| 464 | } | |
| 465 | } | |
| 466 | ||
| 467 | /** | |
| 468 | * Convert any non-standard peptide codes to their standard code table | |
| 469 | * equivalent. (Initial version only does Selenomethionine MSE->MET.) | |
| 470 | * | |
| 471 | * @param threeLetterCode | |
| 472 | * @param seq | |
| 473 | * @param pos | |
| 474 | */ | |
| 475 | 0 | protected void replaceNonCanonicalResidue(String threeLetterCode, |
| 476 | char[] seq, int pos) | |
| 477 | { | |
| 478 | 0 | String canonical = ResidueProperties |
| 479 | .getCanonicalAminoAcid(threeLetterCode); | |
| 480 | 0 | if (canonical != null && !canonical.equalsIgnoreCase(threeLetterCode)) |
| 481 | { | |
| 482 | 0 | seq[pos] = ResidueProperties.getSingleCharacterCode(canonical); |
| 483 | } | |
| 484 | } | |
| 485 | ||
| 486 | /** | |
| 487 | * Not implemented - returns null | |
| 488 | */ | |
| 489 | 0 | @Override |
| 490 | public String print(SequenceI[] seqs, boolean jvSuffix) | |
| 491 | { | |
| 492 | 0 | return null; |
| 493 | } | |
| 494 | ||
| 495 | /** | |
| 496 | * Not implemented | |
| 497 | */ | |
| 498 | 0 | @Override |
| 499 | public void setCallbackFunction(String callbackType, | |
| 500 | String callbackFunction) | |
| 501 | { | |
| 502 | } | |
| 503 | ||
| 504 | 0 | @Override |
| 505 | public void notifyCallback(CBK cbType, Object[] data) | |
| 506 | { | |
| 507 | 0 | String strInfo = (data == null || data[1] == null ? null |
| 508 | : data[1].toString()); | |
| 509 | 0 | switch (cbType) |
| 510 | { | |
| 511 | 0 | case ECHO: |
| 512 | 0 | sendConsoleEcho(strInfo); |
| 513 | 0 | break; |
| 514 | 0 | case SCRIPT: |
| 515 | 0 | notifyScriptTermination((String) data[2], |
| 516 | ((Integer) data[3]).intValue()); | |
| 517 | 0 | break; |
| 518 | 0 | case MEASURE: |
| 519 | 0 | String mystatus = (String) data[3]; |
| 520 | 0 | if (mystatus.indexOf("Picked") >= 0 |
| 521 | || mystatus.indexOf("Sequence") >= 0) | |
| 522 | { | |
| 523 | // Picking mode | |
| 524 | 0 | sendConsoleMessage(strInfo); |
| 525 | } | |
| 526 | 0 | else if (mystatus.indexOf("Completed") >= 0) |
| 527 | { | |
| 528 | 0 | sendConsoleEcho(strInfo.substring(strInfo.lastIndexOf(",") + 2, |
| 529 | strInfo.length() - 1)); | |
| 530 | } | |
| 531 | 0 | break; |
| 532 | 0 | case MESSAGE: |
| 533 | 0 | sendConsoleMessage(data == null ? null : strInfo); |
| 534 | 0 | break; |
| 535 | 0 | case PICK: |
| 536 | 0 | sendConsoleMessage(strInfo); |
| 537 | 0 | break; |
| 538 | 0 | default: |
| 539 | 0 | break; |
| 540 | } | |
| 541 | } | |
| 542 | ||
| 543 | String lastConsoleEcho = ""; | |
| 544 | ||
| 545 | 0 | private void sendConsoleEcho(String string) |
| 546 | { | |
| 547 | 0 | lastConsoleEcho += string; |
| 548 | 0 | lastConsoleEcho += "\n"; |
| 549 | } | |
| 550 | ||
| 551 | String lastConsoleMessage = ""; | |
| 552 | ||
| 553 | 0 | private void sendConsoleMessage(String string) |
| 554 | { | |
| 555 | 0 | lastConsoleMessage += string; |
| 556 | 0 | lastConsoleMessage += "\n"; |
| 557 | } | |
| 558 | ||
| 559 | int lastScriptTermination = -1; | |
| 560 | ||
| 561 | String lastScriptMessage = ""; | |
| 562 | ||
| 563 | 0 | private void notifyScriptTermination(String string, int intValue) |
| 564 | { | |
| 565 | 0 | lastScriptMessage += string; |
| 566 | 0 | lastScriptMessage += "\n"; |
| 567 | 0 | lastScriptTermination = intValue; |
| 568 | } | |
| 569 | ||
| 570 | 0 | @Override |
| 571 | public boolean notifyEnabled(CBK callbackPick) | |
| 572 | { | |
| 573 | 0 | switch (callbackPick) |
| 574 | { | |
| 575 | 0 | case MESSAGE: |
| 576 | 0 | case SCRIPT: |
| 577 | 0 | case ECHO: |
| 578 | 0 | case LOADSTRUCT: |
| 579 | 0 | case ERROR: |
| 580 | 0 | return true; |
| 581 | 0 | default: |
| 582 | 0 | return false; |
| 583 | } | |
| 584 | } | |
| 585 | ||
| 586 | /** | |
| 587 | * Not implemented - returns null | |
| 588 | */ | |
| 589 | 0 | @Override |
| 590 | public String eval(String strEval) | |
| 591 | { | |
| 592 | 0 | return null; |
| 593 | } | |
| 594 | ||
| 595 | /** | |
| 596 | * Not implemented - returns null | |
| 597 | */ | |
| 598 | 0 | @Override |
| 599 | public float[][] functionXY(String functionName, int x, int y) | |
| 600 | { | |
| 601 | 0 | return null; |
| 602 | } | |
| 603 | ||
| 604 | /** | |
| 605 | * Not implemented - returns null | |
| 606 | */ | |
| 607 | 0 | @Override |
| 608 | public float[][][] functionXYZ(String functionName, int nx, int ny, | |
| 609 | int nz) | |
| 610 | { | |
| 611 | 0 | return null; |
| 612 | } | |
| 613 | ||
| 614 | /** | |
| 615 | * Not implemented - returns null | |
| 616 | */ | |
| 617 | 0 | @Override |
| 618 | public String createImage(String fileName, String imageType, | |
| 619 | Object text_or_bytes, int quality) | |
| 620 | { | |
| 621 | 0 | return null; |
| 622 | } | |
| 623 | ||
| 624 | /** | |
| 625 | * Not implemented - returns null | |
| 626 | */ | |
| 627 | 0 | @Override |
| 628 | public Map<String, Object> getRegistryInfo() | |
| 629 | { | |
| 630 | 0 | return null; |
| 631 | } | |
| 632 | ||
| 633 | /** | |
| 634 | * Not implemented | |
| 635 | */ | |
| 636 | 0 | @Override |
| 637 | public void showUrl(String url) | |
| 638 | { | |
| 639 | } | |
| 640 | ||
| 641 | /** | |
| 642 | * Not implemented - returns null | |
| 643 | */ | |
| 644 | 0 | @Override |
| 645 | public int[] resizeInnerPanel(String data) | |
| 646 | { | |
| 647 | 0 | return null; |
| 648 | } | |
| 649 | ||
| 650 | 0 | @Override |
| 651 | public Map<String, Object> getJSpecViewProperty(String arg0) | |
| 652 | { | |
| 653 | 0 | return null; |
| 654 | } | |
| 655 | ||
| 656 | 0 | public boolean isPredictSecondaryStructure() |
| 657 | { | |
| 658 | 0 | return predictSecondaryStructure; |
| 659 | } | |
| 660 | ||
| 661 | 0 | public void setPredictSecondaryStructure( |
| 662 | boolean predictSecondaryStructure) | |
| 663 | { | |
| 664 | 0 | this.predictSecondaryStructure = predictSecondaryStructure; |
| 665 | } | |
| 666 | ||
| 667 | 0 | public boolean isVisibleChainAnnotation() |
| 668 | { | |
| 669 | 0 | return visibleChainAnnotation; |
| 670 | } | |
| 671 | ||
| 672 | 0 | public void setVisibleChainAnnotation(boolean visibleChainAnnotation) |
| 673 | { | |
| 674 | 0 | this.visibleChainAnnotation = visibleChainAnnotation; |
| 675 | } | |
| 676 | ||
| 677 | } |