jalviewX
File PDBfile.java
Coverage histogram
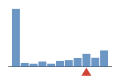
19% of files have more coverage
Classes
| Class | Line # | Actions | ||||
|---|---|---|---|---|---|---|
| PDBfile | 37 | 86 | 34 | 34 |
Contributing tests
This file is covered by 15 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | package mc_view; | |
| 22 | ||
| 23 | import jalview.datamodel.AlignmentAnnotation; | |
| 24 | import jalview.datamodel.DBRefSource; | |
| 25 | import jalview.datamodel.SequenceI; | |
| 26 | import jalview.io.DataSourceType; | |
| 27 | import jalview.io.FileParse; | |
| 28 | import jalview.io.StructureFile; | |
| 29 | import jalview.util.MessageManager; | |
| 30 | ||
| 31 | import java.io.IOException; | |
| 32 | import java.util.ArrayList; | |
| 33 | import java.util.Hashtable; | |
| 34 | import java.util.List; | |
| 35 | import java.util.Vector; | |
| 36 | ||
| 37 | public class PDBfile extends StructureFile | |
| 38 | { | |
| 39 | private static String CALC_ID_PREFIX = "JalviewPDB"; | |
| 40 | ||
| 41 | 0 |  public PDBfile(boolean addAlignmentAnnotations, public PDBfile(boolean addAlignmentAnnotations, |
| 42 | boolean predictSecondaryStructure, boolean externalSecStr) | |
| 43 | { | |
| 44 | 0 | super(); |
| 45 | 0 | addSettings(addAlignmentAnnotations, predictSecondaryStructure, |
| 46 | externalSecStr); | |
| 47 | } | |
| 48 | ||
| 49 | 7 | public PDBfile(boolean addAlignmentAnnotations, boolean predictSecStr, |
| 50 | boolean externalSecStr, String dataObject, | |
| 51 | DataSourceType sourceType) throws IOException | |
| 52 | { | |
| 53 | 7 |  super(false, dataObject, sourceType);
super(false, dataObject, sourceType); |
| 54 | 7 | addSettings(addAlignmentAnnotations, predictSecStr, externalSecStr); |
| 55 | 7 | doParse(); |
| 56 | } | |
| 57 | ||
| 58 | 0 | public PDBfile(boolean addAlignmentAnnotations, boolean predictSecStr, |
| 59 | boolean externalSecStr, FileParse source) throws IOException | |
| 60 | { | |
| 61 | 0 | super(false, source); |
| 62 | 0 | addSettings(addAlignmentAnnotations, predictSecStr, externalSecStr); |
| 63 | 0 | doParse(); |
| 64 | } | |
| 65 | ||
| 66 | 0 | @Override |
| 67 | public String print(SequenceI[] seqs, boolean jvSuffix) | |
| 68 | { | |
| 69 | 0 | return null; |
| 70 | } | |
| 71 | ||
| 72 | 7 | @Override |
| 73 | public void parse() throws IOException | |
| 74 | { | |
| 75 | 7 | setDbRefType(DBRefSource.PDB); |
| 76 | // TODO set the filename sensibly - try using data source name. | |
| 77 | 7 | setId(safeName(getDataName())); |
| 78 | ||
| 79 | 7 | setChains(new Vector<PDBChain>()); |
| 80 | 7 | List<SequenceI> rna = new ArrayList<SequenceI>(); |
| 81 | 7 | List<SequenceI> prot = new ArrayList<SequenceI>(); |
| 82 | 7 | PDBChain tmpchain; |
| 83 | 7 | String line = null; |
| 84 | 7 | boolean modelFlag = false; |
| 85 | 7 | boolean terFlag = false; |
| 86 | 7 | String lastID = ""; |
| 87 | ||
| 88 | 7 | int indexx = 0; |
| 89 | 7 | String atomnam = null; |
| 90 | 7 | try |
| 91 | { | |
| 92 | ? | while ((line = nextLine()) != null) |
| 93 | { | |
| 94 | 29510 | if (line.indexOf("HEADER") == 0) |
| 95 | { | |
| 96 | 7 | if (line.length() > 62) |
| 97 | { | |
| 98 | 7 | String tid; |
| 99 | 7 | if (line.length() > 67) |
| 100 | { | |
| 101 | 5 | tid = line.substring(62, 67).trim(); |
| 102 | } | |
| 103 | else | |
| 104 | { | |
| 105 | 2 | tid = line.substring(62).trim(); |
| 106 | } | |
| 107 | 7 | if (tid.length() > 0) |
| 108 | { | |
| 109 | 7 | setId(tid); |
| 110 | } | |
| 111 | 7 | continue; |
| 112 | } | |
| 113 | } | |
| 114 | // Were we to do anything with SEQRES - we start it here | |
| 115 | 29503 | if (line.indexOf("SEQRES") == 0) |
| 116 | { | |
| 117 | } | |
| 118 | ||
| 119 | 29503 | if (line.indexOf("MODEL") == 0) |
| 120 | { | |
| 121 | 0 | modelFlag = true; |
| 122 | } | |
| 123 | ||
| 124 | 29503 | if (line.indexOf("TER") == 0) |
| 125 | { | |
| 126 | 17 | terFlag = true; |
| 127 | } | |
| 128 | ||
| 129 | 29503 | if (modelFlag && line.indexOf("ENDMDL") == 0) |
| 130 | { | |
| 131 | 0 | break; |
| 132 | } | |
| 133 | 29503 | if (line.indexOf("ATOM") == 0 |
| 134 | || (line.indexOf("HETATM") == 0 && !terFlag)) | |
| 135 | { | |
| 136 | 25446 | terFlag = false; |
| 137 | ||
| 138 | // Jalview is only interested in CA bonds???? | |
| 139 | 25446 | atomnam = line.substring(12, 15).trim(); |
| 140 | 25446 | if (!atomnam.equals("CA") && !atomnam.equals("P")) |
| 141 | { | |
| 142 | 22195 | continue; |
| 143 | } | |
| 144 | ||
| 145 | 3251 | Atom tmpatom = new Atom(line); |
| 146 | 3251 | try |
| 147 | { | |
| 148 | 3251 | tmpchain = findChain(tmpatom.chain); |
| 149 | 3231 | if (tmpatom.resNumIns.trim().equals(lastID)) |
| 150 | { | |
| 151 | // phosphorylated protein - seen both CA and P.. | |
| 152 | 1 | continue; |
| 153 | } | |
| 154 | 3230 | tmpchain.atoms.addElement(tmpatom); |
| 155 | } catch (Exception e) | |
| 156 | { | |
| 157 | 20 | tmpchain = new PDBChain(getId(), tmpatom.chain); |
| 158 | 20 | getChains().add(tmpchain); |
| 159 | 20 | tmpchain.atoms.addElement(tmpatom); |
| 160 | } | |
| 161 | 3250 | lastID = tmpatom.resNumIns.trim(); |
| 162 | } | |
| 163 | 7307 | index++; |
| 164 | } | |
| 165 | ||
| 166 | 7 | makeResidueList(); |
| 167 | 7 | makeCaBondList(); |
| 168 | ||
| 169 | 7 | if (getId() == null) |
| 170 | { | |
| 171 | 0 | setId(inFile.getName()); |
| 172 | } | |
| 173 | 7 | for (PDBChain chain : getChains()) |
| 174 | { | |
| 175 | 20 | SequenceI chainseq = postProcessChain(chain); |
| 176 | 20 | if (isRNA(chainseq)) |
| 177 | { | |
| 178 | 0 | rna.add(chainseq); |
| 179 | } | |
| 180 | else | |
| 181 | { | |
| 182 | 20 | prot.add(chainseq); |
| 183 | } | |
| 184 | } | |
| 185 | 7 | if (predictSecondaryStructure) |
| 186 | { | |
| 187 | 2 | addSecondaryStructure(rna, prot); |
| 188 | } | |
| 189 | } catch (OutOfMemoryError er) | |
| 190 | { | |
| 191 | 0 | System.out.println("OUT OF MEMORY LOADING PDB FILE"); |
| 192 | 0 | throw new IOException(MessageManager |
| 193 | .getString("exception.outofmemory_loading_pdb_file")); | |
| 194 | } catch (NumberFormatException ex) | |
| 195 | { | |
| 196 | 0 | if (line != null) |
| 197 | { | |
| 198 | 0 | System.err.println("Couldn't read number from line:"); |
| 199 | 0 | System.err.println(line); |
| 200 | } | |
| 201 | } | |
| 202 | 7 | markCalcIds(); |
| 203 | } | |
| 204 | ||
| 205 | /** | |
| 206 | * Process a parsed chain to construct and return a Sequence, and add it to | |
| 207 | * the list of sequences parsed. | |
| 208 | * | |
| 209 | * @param chain | |
| 210 | * @return | |
| 211 | */ | |
| 212 | ||
| 213 | 6 | public static boolean isCalcIdHandled(String calcId) |
| 214 | { | |
| 215 | 6 | return calcId != null && (CALC_ID_PREFIX.equals(calcId)); |
| 216 | } | |
| 217 | ||
| 218 | 192 | public static boolean isCalcIdForFile(AlignmentAnnotation alan, |
| 219 | String pdbFile) | |
| 220 | { | |
| 221 | 192 | return alan.getCalcId() != null |
| 222 | && CALC_ID_PREFIX.equals(alan.getCalcId()) | |
| 223 | && pdbFile.equals(alan.getProperty("PDBID")); | |
| 224 | } | |
| 225 | ||
| 226 | 0 | public static String relocateCalcId(String calcId, |
| 227 | Hashtable<String, String> alreadyLoadedPDB) throws Exception | |
| 228 | { | |
| 229 | 0 | int s = CALC_ID_PREFIX.length(), |
| 230 | end = calcId.indexOf(CALC_ID_PREFIX, s); | |
| 231 | 0 | String between = calcId.substring(s, end - 1); |
| 232 | 0 | return CALC_ID_PREFIX + alreadyLoadedPDB.get(between) + ":" |
| 233 | + calcId.substring(end); | |
| 234 | } | |
| 235 | ||
| 236 | 7 | private void markCalcIds() |
| 237 | { | |
| 238 | 7 | for (SequenceI sq : seqs) |
| 239 | { | |
| 240 | 20 | if (sq.getAnnotation() != null) |
| 241 | { | |
| 242 | 12 | for (AlignmentAnnotation aa : sq.getAnnotation()) |
| 243 | { | |
| 244 | 16 | String oldId = aa.getCalcId(); |
| 245 | 16 | if (oldId == null) |
| 246 | { | |
| 247 | 0 | oldId = ""; |
| 248 | } | |
| 249 | 16 | aa.setCalcId(CALC_ID_PREFIX); |
| 250 | 16 | aa.setProperty("PDBID", getId()); |
| 251 | 16 | aa.setProperty("oldCalcId", oldId); |
| 252 | } | |
| 253 | } | |
| 254 | } | |
| 255 | } | |
| 256 | ||
| 257 | } |