Coverage Report
File EnsemblGene.java
Coverage histogram
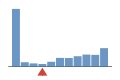
52% of files have more coverage
Classes
| Class | Line # | Actions | |||
|---|---|---|---|---|---|
| EnsemblGene | 52 | 161 | 59 |
Contributing tests
This file is covered by 167 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | package jalview.ext.ensembl; | |
| 22 | ||
| 23 | import jalview.api.FeatureColourI; | |
| 24 | import jalview.api.FeatureSettingsModelI; | |
| 25 | import jalview.datamodel.AlignmentI; | |
| 26 | import jalview.datamodel.GeneLociI; | |
| 27 | import jalview.datamodel.Sequence; | |
| 28 | import jalview.datamodel.SequenceFeature; | |
| 29 | import jalview.datamodel.SequenceI; | |
| 30 | import jalview.datamodel.features.SequenceFeatures; | |
| 31 | import jalview.io.gff.SequenceOntologyFactory; | |
| 32 | import jalview.io.gff.SequenceOntologyI; | |
| 33 | import jalview.schemes.FeatureColour; | |
| 34 | import jalview.schemes.FeatureSettingsAdapter; | |
| 35 | import jalview.util.MapList; | |
| 36 | import jalview.util.Platform; | |
| 37 | ||
| 38 | import java.awt.Color; | |
| 39 | import java.io.UnsupportedEncodingException; | |
| 40 | import java.net.URLDecoder; | |
| 41 | import java.util.ArrayList; | |
| 42 | import java.util.Arrays; | |
| 43 | import java.util.List; | |
| 44 | ||
| 45 | import com.stevesoft.pat.Regex; | |
| 46 | ||
| 47 | /** | |
| 48 | * A class that fetches genomic sequence and all transcripts for an Ensembl gene | |
| 49 | * | |
| 50 | * @author gmcarstairs | |
| 51 | */ | |
| 52 | public class EnsemblGene extends EnsemblSeqProxy | |
| 53 | { | |
| 54 | /* | |
| 55 | * accepts anything as we will attempt lookup of gene or | |
| 56 | * transcript id or gene name | |
| 57 | */ | |
| 58 | private static final Regex ACCESSION_REGEX = new Regex(".*"); | |
| 59 | ||
| 60 | private static final EnsemblFeatureType[] FEATURES_TO_FETCH = { | |
| 61 | EnsemblFeatureType.gene, EnsemblFeatureType.transcript, | |
| 62 | EnsemblFeatureType.exon, EnsemblFeatureType.cds, | |
| 63 | EnsemblFeatureType.variation }; | |
| 64 | ||
| 65 | private static final String CHROMOSOME = "chromosome"; | |
| 66 | ||
| 67 | /** | |
| 68 | * Default constructor (to use rest.ensembl.org) | |
| 69 | */ | |
| 70 | 22 |  public EnsemblGene() public EnsemblGene() |
| 71 | { | |
| 72 | 22 | super(); |
| 73 | } | |
| 74 | ||
| 75 | /** | |
| 76 | * Constructor given the target domain to fetch data from | |
| 77 | * | |
| 78 | * @param d | |
| 79 | */ | |
| 80 | 0 | public EnsemblGene(String d) |
| 81 | { | |
| 82 | 0 | super(d); |
| 83 | } | |
| 84 | ||
| 85 | 2217 | @Override |
| 86 | public String getDbName() | |
| 87 | { | |
| 88 | 2217 | return "ENSEMBL"; |
| 89 | } | |
| 90 | ||
| 91 | 0 | @Override |
| 92 | protected EnsemblFeatureType[] getFeaturesToFetch() | |
| 93 | { | |
| 94 | 0 | return FEATURES_TO_FETCH; |
| 95 | } | |
| 96 | ||
| 97 | 0 | @Override |
| 98 | protected EnsemblSeqType getSourceEnsemblType() | |
| 99 | { | |
| 100 | 0 | return EnsemblSeqType.GENOMIC; |
| 101 | } | |
| 102 | ||
| 103 | 0 | @Override |
| 104 | protected String getObjectType() | |
| 105 | { | |
| 106 | 0 | return OBJECT_TYPE_GENE; |
| 107 | } | |
| 108 | ||
| 109 | /** | |
| 110 | * Returns an alignment containing the gene(s) for the given gene or | |
| 111 | * transcript identifier, or external identifier (e.g. Uniprot id). If given a | |
| 112 | * gene name or external identifier, returns any related gene sequences found | |
| 113 | * for model organisms. If only a single gene is queried for, then its | |
| 114 | * transcripts are also retrieved and added to the alignment. <br> | |
| 115 | * Method: | |
| 116 | * <ul> | |
| 117 | * <li>resolves a transcript identifier by looking up its parent gene id</li> | |
| 118 | * <li>resolves an external identifier by looking up xref-ed gene ids</li> | |
| 119 | * <li>fetches the gene sequence</li> | |
| 120 | * <li>fetches features on the sequence</li> | |
| 121 | * <li>identifies "transcript" features whose Parent is the requested | |
| 122 | * gene</li> | |
| 123 | * <li>fetches the transcript sequence for each transcript</li> | |
| 124 | * <li>makes a mapping from the gene to each transcript</li> | |
| 125 | * <li>copies features from gene to transcript sequences</li> | |
| 126 | * <li>fetches the protein sequence for each transcript, maps and saves it as | |
| 127 | * a cross-reference</li> | |
| 128 | * <li>aligns each transcript against the gene sequence based on the position | |
| 129 | * mappings</li> | |
| 130 | * </ul> | |
| 131 | * | |
| 132 | * @param query | |
| 133 | * a single gene or transcript identifier or gene name | |
| 134 | * @return an alignment containing a gene, and possibly transcripts, or null | |
| 135 | */ | |
| 136 | 0 | @Override |
| 137 | public AlignmentI getSequenceRecords(String query) throws Exception | |
| 138 | { | |
| 139 | /* | |
| 140 | * convert to a non-duplicated list of gene identifiers | |
| 141 | */ | |
| 142 | 0 | List<String> geneIds = getGeneIds(query); |
| 143 | 0 | AlignmentI al = null; |
| 144 | 0 | for (String geneId : geneIds) |
| 145 | { | |
| 146 | /* | |
| 147 | * fetch the gene sequence(s) with features and xrefs | |
| 148 | */ | |
| 149 | 0 | AlignmentI geneAlignment = super.getSequenceRecords(geneId); |
| 150 | 0 | if (geneAlignment == null) |
| 151 | { | |
| 152 | 0 | continue; |
| 153 | } | |
| 154 | ||
| 155 | 0 | if (geneAlignment.getHeight() == 1) |
| 156 | { | |
| 157 | // ensure id has 'correct' case for the Ensembl identifier | |
| 158 | 0 | geneId = geneAlignment.getSequenceAt(0).getName(); |
| 159 | 0 | findGeneLoci(geneAlignment.getSequenceAt(0), geneId); |
| 160 | 0 | getTranscripts(geneAlignment, geneId); |
| 161 | } | |
| 162 | 0 | if (al == null) |
| 163 | { | |
| 164 | 0 | al = geneAlignment; |
| 165 | } | |
| 166 | else | |
| 167 | { | |
| 168 | 0 | al.append(geneAlignment); |
| 169 | } | |
| 170 | } | |
| 171 | 0 | return al; |
| 172 | } | |
| 173 | ||
| 174 | /** | |
| 175 | * Calls the /lookup/id REST service, parses the response for gene | |
| 176 | * coordinates, and if successful, adds these to the sequence. If this fails, | |
| 177 | * fall back on trying to parse the sequence description in case it is in | |
| 178 | * Ensembl-gene format e.g. chromosome:GRCh38:17:45051610:45109016:1. | |
| 179 | * | |
| 180 | * @param seq | |
| 181 | * @param geneId | |
| 182 | */ | |
| 183 | 0 | void findGeneLoci(SequenceI seq, String geneId) |
| 184 | { | |
| 185 | 0 | GeneLociI geneLoci = new EnsemblLookup(getDomain()).getGeneLoci(geneId); |
| 186 | 0 | if (geneLoci != null) |
| 187 | { | |
| 188 | 0 | seq.setGeneLoci(geneLoci.getSpeciesId(), geneLoci.getAssemblyId(), |
| 189 | geneLoci.getChromosomeId(), geneLoci.getMapping()); | |
| 190 | } | |
| 191 | else | |
| 192 | { | |
| 193 | 0 | parseChromosomeLocations(seq); |
| 194 | } | |
| 195 | } | |
| 196 | ||
| 197 | /** | |
| 198 | * Parses and saves fields of an Ensembl-style description e.g. | |
| 199 | * chromosome:GRCh38:17:45051610:45109016:1 | |
| 200 | * | |
| 201 | * @param seq | |
| 202 | */ | |
| 203 | 0 | boolean parseChromosomeLocations(SequenceI seq) |
| 204 | { | |
| 205 | 0 | String description = seq.getDescription(); |
| 206 | 0 | if (description == null) |
| 207 | { | |
| 208 | 0 | return false; |
| 209 | } | |
| 210 | 0 | String[] tokens = description.split(":"); |
| 211 | 0 | if (tokens.length == 6 && tokens[0].startsWith(CHROMOSOME)) |
| 212 | { | |
| 213 | 0 | String ref = tokens[1]; |
| 214 | 0 | String chrom = tokens[2]; |
| 215 | 0 | try |
| 216 | { | |
| 217 | 0 | int chStart = Integer.parseInt(tokens[3]); |
| 218 | 0 | int chEnd = Integer.parseInt(tokens[4]); |
| 219 | 0 | boolean forwardStrand = "1".equals(tokens[5]); |
| 220 | 0 | String species = ""; // not known here |
| 221 | 0 | int[] from = new int[] { seq.getStart(), seq.getEnd() }; |
| 222 | 0 | int[] to = new int[] { forwardStrand ? chStart : chEnd, |
| 223 | 0 | forwardStrand ? chEnd : chStart }; |
| 224 | 0 | MapList map = new MapList(from, to, 1, 1); |
| 225 | 0 | seq.setGeneLoci(species, ref, chrom, map); |
| 226 | 0 | return true; |
| 227 | } catch (NumberFormatException e) | |
| 228 | { | |
| 229 | 0 | jalview.bin.Console |
| 230 | .errPrintln("Bad integers in description " + description); | |
| 231 | } | |
| 232 | } | |
| 233 | 0 | return false; |
| 234 | } | |
| 235 | ||
| 236 | /** | |
| 237 | * Converts a query, which may contain one or more gene, transcript, or | |
| 238 | * external (to Ensembl) identifiers, into a non-redundant list of gene | |
| 239 | * identifiers. | |
| 240 | * | |
| 241 | * @param accessions | |
| 242 | * @return | |
| 243 | */ | |
| 244 | 0 | List<String> getGeneIds(String accessions) |
| 245 | { | |
| 246 | 0 | List<String> geneIds = new ArrayList<>(); |
| 247 | ||
| 248 | 0 | for (String acc : accessions.split(getAccessionSeparator())) |
| 249 | { | |
| 250 | /* | |
| 251 | * First try lookup as an Ensembl (gene or transcript) identifier | |
| 252 | */ | |
| 253 | 0 | String geneId = new EnsemblLookup(getDomain()).getGeneId(acc); |
| 254 | 0 | if (geneId != null) |
| 255 | { | |
| 256 | 0 | if (!geneIds.contains(geneId)) |
| 257 | { | |
| 258 | 0 | geneIds.add(geneId); |
| 259 | } | |
| 260 | } | |
| 261 | else | |
| 262 | { | |
| 263 | /* | |
| 264 | * if given a gene or other external name, lookup and fetch | |
| 265 | * the corresponding gene for all model organisms | |
| 266 | */ | |
| 267 | 0 | List<String> ids = new EnsemblSymbol(getDomain(), getDbSource(), |
| 268 | getDbVersion()).getGeneIds(acc); | |
| 269 | 0 | for (String id : ids) |
| 270 | { | |
| 271 | 0 | if (!geneIds.contains(id)) |
| 272 | { | |
| 273 | 0 | geneIds.add(id); |
| 274 | } | |
| 275 | } | |
| 276 | } | |
| 277 | } | |
| 278 | 0 | return geneIds; |
| 279 | } | |
| 280 | ||
| 281 | /** | |
| 282 | * Constructs all transcripts for the gene, as identified by "transcript" | |
| 283 | * features whose Parent is the requested gene. The coding transcript | |
| 284 | * sequences (i.e. with introns omitted) are added to the alignment. | |
| 285 | * | |
| 286 | * @param al | |
| 287 | * @param accId | |
| 288 | * @throws Exception | |
| 289 | */ | |
| 290 | 0 | protected void getTranscripts(AlignmentI al, String accId) |
| 291 | throws Exception | |
| 292 | { | |
| 293 | 0 | SequenceI gene = al.getSequenceAt(0); |
| 294 | 0 | List<SequenceFeature> transcriptFeatures = getTranscriptFeatures(accId, |
| 295 | gene); | |
| 296 | ||
| 297 | 0 | for (SequenceFeature transcriptFeature : transcriptFeatures) |
| 298 | { | |
| 299 | 0 | makeTranscript(transcriptFeature, al, gene); |
| 300 | } | |
| 301 | ||
| 302 | 0 | clearGeneFeatures(gene); |
| 303 | } | |
| 304 | ||
| 305 | /** | |
| 306 | * Remove unwanted features (transcript, exon, CDS) from the gene sequence | |
| 307 | * after we have used them to derive transcripts and transfer features | |
| 308 | * | |
| 309 | * @param gene | |
| 310 | */ | |
| 311 | 0 | protected void clearGeneFeatures(SequenceI gene) |
| 312 | { | |
| 313 | /* | |
| 314 | * Note we include NMD_transcript_variant here because it behaves like | |
| 315 | * 'transcript' in Ensembl, although strictly speaking it is not | |
| 316 | * (it is a sub-type of sequence_variant) | |
| 317 | */ | |
| 318 | 0 | String[] soTerms = new String[] { |
| 319 | SequenceOntologyI.NMD_TRANSCRIPT_VARIANT, | |
| 320 | SequenceOntologyI.TRANSCRIPT, SequenceOntologyI.EXON, | |
| 321 | SequenceOntologyI.CDS }; | |
| 322 | 0 | List<SequenceFeature> sfs = gene.getFeatures() |
| 323 | .getFeaturesByOntology(soTerms); | |
| 324 | 0 | for (SequenceFeature sf : sfs) |
| 325 | { | |
| 326 | 0 | gene.deleteFeature(sf); |
| 327 | } | |
| 328 | } | |
| 329 | ||
| 330 | /** | |
| 331 | * Constructs a spliced transcript sequence by finding 'exon' features for the | |
| 332 | * given id (or failing that 'CDS'). Copies features on to the new sequence. | |
| 333 | * 'Aligns' the new sequence against the gene sequence by padding with gaps, | |
| 334 | * and adds it to the alignment. | |
| 335 | * | |
| 336 | * @param transcriptFeature | |
| 337 | * @param al | |
| 338 | * the alignment to which to add the new sequence | |
| 339 | * @param gene | |
| 340 | * the parent gene sequence, with features | |
| 341 | * @return | |
| 342 | */ | |
| 343 | 0 | SequenceI makeTranscript(SequenceFeature transcriptFeature, AlignmentI al, |
| 344 | SequenceI gene) | |
| 345 | { | |
| 346 | 0 | String accId = getTranscriptId(transcriptFeature); |
| 347 | 0 | if (accId == null) |
| 348 | { | |
| 349 | 0 | return null; |
| 350 | } | |
| 351 | ||
| 352 | /* | |
| 353 | * NB we are mapping from gene sequence (not genome), so do not | |
| 354 | * need to check for reverse strand (gene and transcript sequences | |
| 355 | * are in forward sense) | |
| 356 | */ | |
| 357 | ||
| 358 | /* | |
| 359 | * make a gene-length sequence filled with gaps | |
| 360 | * we will fill in the bases for transcript regions | |
| 361 | */ | |
| 362 | 0 | char[] seqChars = new char[gene.getLength()]; |
| 363 | 0 | Arrays.fill(seqChars, al.getGapCharacter()); |
| 364 | ||
| 365 | /* | |
| 366 | * look for exon features of the transcript, failing that for CDS | |
| 367 | * (for example ENSG00000124610 has 1 CDS but no exon features) | |
| 368 | */ | |
| 369 | 0 | String parentId = accId; |
| 370 | 0 | List<SequenceFeature> splices = findFeatures(gene, |
| 371 | SequenceOntologyI.EXON, parentId); | |
| 372 | 0 | if (splices.isEmpty()) |
| 373 | { | |
| 374 | 0 | splices = findFeatures(gene, SequenceOntologyI.CDS, parentId); |
| 375 | } | |
| 376 | 0 | SequenceFeatures.sortFeatures(splices, true); |
| 377 | ||
| 378 | 0 | int transcriptLength = 0; |
| 379 | 0 | final char[] geneChars = gene.getSequence(); |
| 380 | 0 | int offset = gene.getStart(); // to convert to 0-based positions |
| 381 | 0 | List<int[]> mappedFrom = new ArrayList<>(); |
| 382 | ||
| 383 | 0 | for (SequenceFeature sf : splices) |
| 384 | { | |
| 385 | 0 | int start = sf.getBegin() - offset; |
| 386 | 0 | int end = sf.getEnd() - offset; |
| 387 | 0 | int spliceLength = end - start + 1; |
| 388 | 0 | System.arraycopy(geneChars, start, seqChars, start, spliceLength); |
| 389 | 0 | transcriptLength += spliceLength; |
| 390 | 0 | mappedFrom.add(new int[] { sf.getBegin(), sf.getEnd() }); |
| 391 | } | |
| 392 | ||
| 393 | 0 | Sequence transcript = new Sequence(accId, seqChars, 1, |
| 394 | transcriptLength); | |
| 395 | ||
| 396 | /* | |
| 397 | * Ensembl has gene name as transcript Name | |
| 398 | * EnsemblGenomes doesn't, but has a url-encoded description field | |
| 399 | */ | |
| 400 | 0 | String description = transcriptFeature.getDescription(); |
| 401 | 0 | if (description == null) |
| 402 | { | |
| 403 | 0 | description = (String) transcriptFeature.getValue(DESCRIPTION); |
| 404 | } | |
| 405 | 0 | if (description != null) |
| 406 | { | |
| 407 | 0 | try |
| 408 | { | |
| 409 | 0 | transcript.setDescription(URLDecoder.decode(description, "UTF-8")); |
| 410 | } catch (UnsupportedEncodingException e) | |
| 411 | { | |
| 412 | 0 | e.printStackTrace(); // as if |
| 413 | } | |
| 414 | } | |
| 415 | 0 | transcript.createDatasetSequence(); |
| 416 | ||
| 417 | 0 | al.addSequence(transcript); |
| 418 | ||
| 419 | /* | |
| 420 | * transfer features to the new sequence; we use EnsemblCdna to do this, | |
| 421 | * to filter out unwanted features types (see method retainFeature) | |
| 422 | */ | |
| 423 | 0 | List<int[]> mapTo = new ArrayList<>(); |
| 424 | 0 | mapTo.add(new int[] { 1, transcriptLength }); |
| 425 | 0 | MapList mapping = new MapList(mappedFrom, mapTo, 1, 1); |
| 426 | 0 | EnsemblCdna cdna = new EnsemblCdna(getDomain()); |
| 427 | 0 | cdna.transferFeatures(gene.getFeatures().getPositionalFeatures(), |
| 428 | transcript.getDatasetSequence(), mapping, parentId); | |
| 429 | ||
| 430 | 0 | mapTranscriptToChromosome(transcript, gene, mapping); |
| 431 | ||
| 432 | /* | |
| 433 | * fetch and save cross-references | |
| 434 | */ | |
| 435 | 0 | cdna.getCrossReferences(transcript); |
| 436 | ||
| 437 | /* | |
| 438 | * and finally fetch the protein product and save as a cross-reference | |
| 439 | */ | |
| 440 | 0 | cdna.addProteinProduct(transcript); |
| 441 | ||
| 442 | 0 | return transcript; |
| 443 | } | |
| 444 | ||
| 445 | /** | |
| 446 | * If the gene has a mapping to chromosome coordinates, derive the transcript | |
| 447 | * chromosome regions and save on the transcript sequence | |
| 448 | * | |
| 449 | * @param transcript | |
| 450 | * @param gene | |
| 451 | * @param mapping | |
| 452 | * the mapping from gene to transcript positions | |
| 453 | */ | |
| 454 | 0 | protected void mapTranscriptToChromosome(SequenceI transcript, |
| 455 | SequenceI gene, MapList mapping) | |
| 456 | { | |
| 457 | 0 | GeneLociI loci = gene.getGeneLoci(); |
| 458 | 0 | if (loci == null) |
| 459 | { | |
| 460 | 0 | return; |
| 461 | } | |
| 462 | ||
| 463 | 0 | MapList geneMapping = loci.getMapping(); |
| 464 | ||
| 465 | 0 | List<int[]> exons = mapping.getFromRanges(); |
| 466 | 0 | List<int[]> transcriptLoci = new ArrayList<>(); |
| 467 | ||
| 468 | 0 | for (int[] exon : exons) |
| 469 | { | |
| 470 | 0 | transcriptLoci.add(geneMapping.locateInTo(exon[0], exon[1])); |
| 471 | } | |
| 472 | ||
| 473 | 0 | List<int[]> transcriptRange = Arrays |
| 474 | .asList(new int[] | |
| 475 | { transcript.getStart(), transcript.getEnd() }); | |
| 476 | 0 | MapList mapList = new MapList(transcriptRange, transcriptLoci, 1, 1); |
| 477 | ||
| 478 | 0 | transcript.setGeneLoci(loci.getSpeciesId(), loci.getAssemblyId(), |
| 479 | loci.getChromosomeId(), mapList); | |
| 480 | } | |
| 481 | ||
| 482 | /** | |
| 483 | * Returns the 'transcript_id' property of the sequence feature (or null) | |
| 484 | * | |
| 485 | * @param feature | |
| 486 | * @return | |
| 487 | */ | |
| 488 | 0 | protected String getTranscriptId(SequenceFeature feature) |
| 489 | { | |
| 490 | 0 | return (String) feature.getValue(JSON_ID); |
| 491 | } | |
| 492 | ||
| 493 | /** | |
| 494 | * Returns a list of the transcript features on the sequence whose Parent is | |
| 495 | * the gene for the accession id. | |
| 496 | * <p> | |
| 497 | * Transcript features are those of type "transcript", or any of its sub-types | |
| 498 | * in the Sequence Ontology e.g. "mRNA", "processed_transcript". We also | |
| 499 | * include "NMD_transcript_variant", because this type behaves like a | |
| 500 | * transcript identifier in Ensembl, although strictly speaking it is not in | |
| 501 | * the SO. | |
| 502 | * | |
| 503 | * @param accId | |
| 504 | * @param geneSequence | |
| 505 | * @return | |
| 506 | */ | |
| 507 | 1 | protected List<SequenceFeature> getTranscriptFeatures(String accId, |
| 508 | SequenceI geneSequence) | |
| 509 | { | |
| 510 | 1 | List<SequenceFeature> transcriptFeatures = new ArrayList<>(); |
| 511 | ||
| 512 | 1 | String parentIdentifier = accId; |
| 513 | ||
| 514 | 1 | List<SequenceFeature> sfs = geneSequence.getFeatures() |
| 515 | .getFeaturesByOntology(SequenceOntologyI.TRANSCRIPT); | |
| 516 | 1 | sfs.addAll(geneSequence.getFeatures().getPositionalFeatures( |
| 517 | SequenceOntologyI.NMD_TRANSCRIPT_VARIANT)); | |
| 518 | ||
| 519 | 1 | for (SequenceFeature sf : sfs) |
| 520 | { | |
| 521 | 4 | String parent = (String) sf.getValue(PARENT); |
| 522 | 4 | if (parentIdentifier.equalsIgnoreCase(parent)) |
| 523 | { | |
| 524 | 3 | transcriptFeatures.add(sf); |
| 525 | } | |
| 526 | } | |
| 527 | ||
| 528 | 1 | return transcriptFeatures; |
| 529 | } | |
| 530 | ||
| 531 | 0 | @Override |
| 532 | public String getDescription() | |
| 533 | { | |
| 534 | 0 | return "Fetches all transcripts and variant features for a gene or transcript"; |
| 535 | } | |
| 536 | ||
| 537 | /** | |
| 538 | * Default test query is a gene id (can also enter a transcript id) | |
| 539 | */ | |
| 540 | 0 | @Override |
| 541 | public String getTestQuery() | |
| 542 | { | |
| 543 | 0 | return Platform.isJS() ? "ENSG00000123569" : "ENSG00000157764"; |
| 544 | // ENSG00000123569 // H2BFWT histone, 2 transcripts, reverse strand | |
| 545 | // ENSG00000157764 // BRAF, 5 transcripts, reverse strand | |
| 546 | // ENSG00000090266 // NDUFB2, 15 transcripts, forward strand | |
| 547 | // ENSG00000101812 // H2BFM histone, 3 transcripts, forward strand | |
| 548 | } | |
| 549 | ||
| 550 | /** | |
| 551 | * Answers a list of sequence features (if any) whose type is 'gene' (or a | |
| 552 | * subtype of gene in the Sequence Ontology), and whose ID is the accession we | |
| 553 | * are retrieving | |
| 554 | */ | |
| 555 | 3 | @Override |
| 556 | protected List<SequenceFeature> getIdentifyingFeatures(SequenceI seq, | |
| 557 | String accId) | |
| 558 | { | |
| 559 | 3 | List<SequenceFeature> result = new ArrayList<>(); |
| 560 | 3 | List<SequenceFeature> sfs = seq.getFeatures() |
| 561 | .getFeaturesByOntology(SequenceOntologyI.GENE); | |
| 562 | 3 | for (SequenceFeature sf : sfs) |
| 563 | { | |
| 564 | 6 | String id = (String) sf.getValue(JSON_ID); |
| 565 | 6 | if (accId.equalsIgnoreCase(id)) |
| 566 | { | |
| 567 | 4 | result.add(sf); |
| 568 | } | |
| 569 | } | |
| 570 | 3 | return result; |
| 571 | } | |
| 572 | ||
| 573 | /** | |
| 574 | * Answers true unless feature type is 'gene', or 'transcript' with a parent | |
| 575 | * which is a different gene. We need the gene features to identify the range, | |
| 576 | * but it is redundant information on the gene sequence. Checking the parent | |
| 577 | * allows us to drop transcript features which belong to different | |
| 578 | * (overlapping) genes. | |
| 579 | */ | |
| 580 | 6 | @Override |
| 581 | protected boolean retainFeature(SequenceFeature sf, String accessionId) | |
| 582 | { | |
| 583 | 6 | SequenceOntologyI so = SequenceOntologyFactory.getInstance(); |
| 584 | 6 | String type = sf.getType(); |
| 585 | 6 | if (so.isA(type, SequenceOntologyI.GENE)) |
| 586 | { | |
| 587 | 1 | return false; |
| 588 | } | |
| 589 | 5 | if (isTranscript(type)) |
| 590 | { | |
| 591 | 4 | String parent = (String) sf.getValue(PARENT); |
| 592 | 4 | if (!accessionId.equalsIgnoreCase(parent)) |
| 593 | { | |
| 594 | 1 | return false; |
| 595 | } | |
| 596 | } | |
| 597 | 4 | return true; |
| 598 | } | |
| 599 | ||
| 600 | /** | |
| 601 | * Override to do nothing as Ensembl doesn't return a protein sequence for a | |
| 602 | * gene identifier | |
| 603 | */ | |
| 604 | 0 | @Override |
| 605 | protected void addProteinProduct(SequenceI querySeq) | |
| 606 | { | |
| 607 | } | |
| 608 | ||
| 609 | 0 | @Override |
| 610 | public Regex getAccessionValidator() | |
| 611 | { | |
| 612 | 0 | return ACCESSION_REGEX; |
| 613 | } | |
| 614 | ||
| 615 | /** | |
| 616 | * Returns a descriptor for suitable feature display settings with | |
| 617 | * <ul> | |
| 618 | * <li>only exon or sequence_variant features (or their subtypes in the | |
| 619 | * Sequence Ontology) visible</li> | |
| 620 | * <li>variant features coloured red</li> | |
| 621 | * <li>exon features coloured by label (exon name)</li> | |
| 622 | * <li>variants displayed above (on top of) exons</li> | |
| 623 | * </ul> | |
| 624 | */ | |
| 625 | 3 | @Override |
| 626 | public FeatureSettingsModelI getFeatureColourScheme() | |
| 627 | { | |
| 628 | 3 | return new FeatureSettingsAdapter() |
| 629 | { | |
| 630 | SequenceOntologyI so = SequenceOntologyFactory.getInstance(); | |
| 631 | ||
| 632 | 20 | @Override |
| 633 | public boolean isFeatureHidden(String type) | |
| 634 | { | |
| 635 | 20 | return (!so.isA(type, SequenceOntologyI.EXON) |
| 636 | && !so.isA(type, SequenceOntologyI.SEQUENCE_VARIANT)); | |
| 637 | } | |
| 638 | ||
| 639 | 22 | @Override |
| 640 | public FeatureColourI getFeatureColour(String type) | |
| 641 | { | |
| 642 | 22 | if (so.isA(type, SequenceOntologyI.EXON)) |
| 643 | { | |
| 644 | 8 | return new FeatureColour() |
| 645 | { | |
| 646 | 2 | @Override |
| 647 | public boolean isColourByLabel() | |
| 648 | { | |
| 649 | 2 | return true; |
| 650 | } | |
| 651 | }; | |
| 652 | } | |
| 653 | 14 | if (so.isA(type, SequenceOntologyI.SEQUENCE_VARIANT)) |
| 654 | { | |
| 655 | 12 | return new FeatureColour() |
| 656 | { | |
| 657 | ||
| 658 | 2 | @Override |
| 659 | public Color getColour() | |
| 660 | { | |
| 661 | 2 | return Color.RED; |
| 662 | } | |
| 663 | }; | |
| 664 | } | |
| 665 | 2 | return null; |
| 666 | } | |
| 667 | ||
| 668 | /** | |
| 669 | * order to render sequence_variant after exon after the rest | |
| 670 | */ | |
| 671 | 16 | @Override |
| 672 | public int compare(String feature1, String feature2) | |
| 673 | { | |
| 674 | 16 | if (so.isA(feature1, SequenceOntologyI.SEQUENCE_VARIANT)) |
| 675 | { | |
| 676 | 12 | return +1; |
| 677 | } | |
| 678 | 4 | if (so.isA(feature2, SequenceOntologyI.SEQUENCE_VARIANT)) |
| 679 | { | |
| 680 | 2 | return -1; |
| 681 | } | |
| 682 | 2 | if (so.isA(feature1, SequenceOntologyI.EXON)) |
| 683 | { | |
| 684 | 2 | return +1; |
| 685 | } | |
| 686 | 0 | if (so.isA(feature2, SequenceOntologyI.EXON)) |
| 687 | { | |
| 688 | 0 | return -1; |
| 689 | } | |
| 690 | 0 | return 0; |
| 691 | } | |
| 692 | }; | |
| 693 | } | |
| 694 | ||
| 695 | } |