jalviewX
File SimilarityParams.java
Coverage histogram
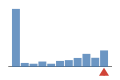
0% of files have more coverage
Classes
| Class | Line # | Actions | ||||
|---|---|---|---|---|---|---|
| SimilarityParams | 37 | 8 | 5 | 0 |
Contributing tests
This file is covered by 8 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | package jalview.analysis.scoremodels; | |
| 22 | ||
| 23 | import jalview.api.analysis.SimilarityParamsI; | |
| 24 | ||
| 25 | /** | |
| 26 | * A class to hold parameters that configure the pairwise similarity | |
| 27 | * calculation. Based on the paper | |
| 28 | * | |
| 29 | * <pre> | |
| 30 | * Quantification of the variation in percentage identity for protein sequence alignments | |
| 31 | * Raghava, GP and Barton, GJ | |
| 32 | * BMC Bioinformatics. 2006 Sep 19;7:415 | |
| 33 | * </pre> | |
| 34 | * | |
| 35 | * @see https://www.ncbi.nlm.nih.gov/pubmed/16984632 | |
| 36 | */ | |
| 37 | public class SimilarityParams implements SimilarityParamsI | |
| 38 | { | |
| 39 | /** | |
| 40 | * Based on Jalview's Comparison.PID method, which includes gaps and counts | |
| 41 | * them as matching; it counts over the length of the shorter sequence | |
| 42 | */ | |
| 43 | public static final SimilarityParamsI Jalview = new SimilarityParams(true, | |
| 44 | true, true, true); | |
| 45 | ||
| 46 | /** | |
| 47 | * 'SeqSpace' mode PCA calculation includes gaps but does not count them as | |
| 48 | * matching; it uses the longest sequence length | |
| 49 | */ | |
| 50 | public static final SimilarityParamsI SeqSpace = new SimilarityParams( | |
| 51 | true, false, true, true); | |
| 52 | ||
| 53 | /** | |
| 54 | * as described in the Raghava-Barton paper | |
| 55 | * <ul> | |
| 56 | * <li>ignores gap-gap</li> | |
| 57 | * <li>does not score gap-residue</li> | |
| 58 | * <li>includes gap-residue in lengths</li> | |
| 59 | * <li>matches on longer of two sequences</li> | |
| 60 | * </ul> | |
| 61 | */ | |
| 62 | public static final SimilarityParamsI PID1 = new SimilarityParams(false, | |
| 63 | false, true, false); | |
| 64 | ||
| 65 | /** | |
| 66 | * as described in the Raghava-Barton paper | |
| 67 | * <ul> | |
| 68 | * <li>ignores gap-gap</li> | |
| 69 | * <li>ignores gap-residue</li> | |
| 70 | * <li>matches on longer of two sequences</li> | |
| 71 | * </ul> | |
| 72 | */ | |
| 73 | public static final SimilarityParamsI PID2 = new SimilarityParams(false, | |
| 74 | false, false, false); | |
| 75 | ||
| 76 | /** | |
| 77 | * as described in the Raghava-Barton paper | |
| 78 | * <ul> | |
| 79 | * <li>ignores gap-gap</li> | |
| 80 | * <li>ignores gap-residue</li> | |
| 81 | * <li>matches on shorter of sequences only</li> | |
| 82 | * </ul> | |
| 83 | */ | |
| 84 | public static final SimilarityParamsI PID3 = new SimilarityParams(false, | |
| 85 | false, false, true); | |
| 86 | ||
| 87 | /** | |
| 88 | * as described in the Raghava-Barton paper | |
| 89 | * <ul> | |
| 90 | * <li>ignores gap-gap</li> | |
| 91 | * <li>does not score gap-residue</li> | |
| 92 | * <li>includes gap-residue in lengths</li> | |
| 93 | * <li>matches on shorter of sequences only</li> | |
| 94 | * </ul> | |
| 95 | */ | |
| 96 | public static final SimilarityParamsI PID4 = new SimilarityParams(false, | |
| 97 | false, true, true); | |
| 98 | ||
| 99 | private boolean includeGappedColumns; | |
| 100 | ||
| 101 | private boolean matchGaps; | |
| 102 | ||
| 103 | private boolean includeGaps; | |
| 104 | ||
| 105 | private boolean denominateByShortestLength; | |
| 106 | ||
| 107 | /** | |
| 108 | * Constructor | |
| 109 | * | |
| 110 | * @param includeGapGap | |
| 111 | * @param matchGapResidue | |
| 112 | * @param includeGapResidue | |
| 113 | * if true, gapped positions are counted for normalisation by length | |
| 114 | * @param shortestLength | |
| 115 | * if true, the denominator is the shorter sequence length (possibly | |
| 116 | * including gaps) | |
| 117 | */ | |
| 118 | 33 |  public SimilarityParams(boolean includeGapGap, boolean matchGapResidue, public SimilarityParams(boolean includeGapGap, boolean matchGapResidue, |
| 119 | boolean includeGapResidue, boolean shortestLength) | |
| 120 | { | |
| 121 | 33 | includeGappedColumns = includeGapGap; |
| 122 | 33 | matchGaps = matchGapResidue; |
| 123 | 33 | includeGaps = includeGapResidue; |
| 124 | 33 | denominateByShortestLength = shortestLength; |
| 125 | } | |
| 126 | ||
| 127 | 82 | @Override |
| 128 | public boolean includeGaps() | |
| 129 | { | |
| 130 | 82 | return includeGaps; |
| 131 | } | |
| 132 | ||
| 133 | 27 | @Override |
| 134 | public boolean denominateByShortestLength() | |
| 135 | { | |
| 136 | 27 | return denominateByShortestLength; |
| 137 | } | |
| 138 | ||
| 139 | 27 | @Override |
| 140 | public boolean includeGappedColumns() | |
| 141 | { | |
| 142 | 27 | return includeGappedColumns; |
| 143 | } | |
| 144 | ||
| 145 | 28 | @Override |
| 146 | public boolean matchGaps() | |
| 147 | { | |
| 148 | 28 | return matchGaps; |
| 149 | } | |
| 150 | } |