Coverage Report
File AnnotationJob.java
Coverage histogram
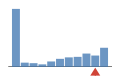
13% of files have more coverage
Classes
| Class | Line # | Actions | |||
|---|---|---|---|---|---|
| AnnotationJob | 19 | 58 | 20 |
Contributing tests
This file is covered by 23 tests.
.
Source view
| 1 | package jalview.ws2.actions.annotation; | |
| 2 | ||
| 3 | import java.util.ArrayList; | |
| 4 | import java.util.Arrays; | |
| 5 | import java.util.BitSet; | |
| 6 | import java.util.HashMap; | |
| 7 | import java.util.List; | |
| 8 | import java.util.Map; | |
| 9 | ||
| 10 | import jalview.analysis.AlignSeq; | |
| 11 | import jalview.analysis.SeqsetUtils; | |
| 12 | import jalview.datamodel.Sequence; | |
| 13 | import jalview.datamodel.SequenceCollectionI; | |
| 14 | import jalview.datamodel.SequenceI; | |
| 15 | import jalview.schemes.ResidueProperties; | |
| 16 | import jalview.util.Comparison; | |
| 17 | import jalview.ws2.actions.BaseJob; | |
| 18 | ||
| 19 | public class AnnotationJob extends BaseJob | |
| 20 | { | |
| 21 | final boolean[] gapMap; | |
| 22 | ||
| 23 | final Map<String, SequenceI> seqNames; | |
| 24 | ||
| 25 | final int regionStart, regionEnd; | |
| 26 | ||
| 27 | final int minSize; | |
| 28 | ||
| 29 | 24 |  public AnnotationJob(List<SequenceI> inputSeqs, boolean[] gapMap, public AnnotationJob(List<SequenceI> inputSeqs, boolean[] gapMap, |
| 30 | Map<String, SequenceI> seqNames, int start, int end, int minSize) | |
| 31 | { | |
| 32 | 24 | super(inputSeqs); |
| 33 | 24 | this.gapMap = gapMap; |
| 34 | 24 | this.seqNames = seqNames; |
| 35 | 24 | this.regionStart = start; |
| 36 | 24 | this.regionEnd = end; |
| 37 | 24 | this.minSize = minSize; |
| 38 | } | |
| 39 | ||
| 40 | 0 | @Override |
| 41 | public boolean isInputValid() | |
| 42 | { | |
| 43 | 0 | int nvalid = 0; |
| 44 | 0 | for (SequenceI sq : getInputSequences()) |
| 45 | 0 | if (sq.getStart() <= sq.getEnd()) |
| 46 | 0 | nvalid++; |
| 47 | 0 | return nvalid >= minSize; |
| 48 | } | |
| 49 | ||
| 50 | 24 | public static AnnotationJob create(SequenceCollectionI inputSeqs, |
| 51 | boolean bySequence, boolean submitGaps, boolean requireAligned, | |
| 52 | boolean filterNonStandardResidues, int minSize) | |
| 53 | { | |
| 54 | 24 | List<SequenceI> sequences = new ArrayList<>(); |
| 55 | 24 | int minlen = 10; |
| 56 | 24 | int width = 0; |
| 57 | 24 | Map<String, SequenceI> namesMap = bySequence ? new HashMap<>() : null; |
| 58 | 24 | BitSet residueMap = new BitSet(); |
| 59 | 24 | int start = inputSeqs.getStartRes(); |
| 60 | 24 | int end = inputSeqs.getEndRes(); |
| 61 | // TODO: URGENT! unify with JPred / MSA code to handle hidden regions | |
| 62 | // correctly | |
| 63 | // TODO: push attributes into WsJob instance (so they can be safely | |
| 64 | // persisted/restored | |
| 65 | 24 | for (SequenceI sq : inputSeqs.getSequences()) |
| 66 | { | |
| 67 | 64 | int sqLen = (bySequence) |
| 68 | ? sq.findPosition(end + 1) - sq.findPosition(start + 1) | |
| 69 | : sq.getEnd() - sq.getStart(); | |
| 70 | 64 | if (sqLen < minlen) |
| 71 | 18 | continue; |
| 72 | 46 | width = Math.max(width, sq.getLength()); |
| 73 | 46 | String newName = SeqsetUtils.unique_name(sequences.size() + 1); |
| 74 | 46 | if (namesMap != null) |
| 75 | 46 | namesMap.put(newName, sq); |
| 76 | 46 | char[] seqChars = sq.getSequence(start, end + 1); |
| 77 | 46 | if (filterNonStandardResidues) |
| 78 | 8 | replaceNonStandardResidues(seqChars, Comparison.GAP_DASH, |
| 79 | sq.isProtein()); | |
| 80 | 46 | Sequence seq; |
| 81 | 46 | if (submitGaps) |
| 82 | { | |
| 83 | 33 | seq = new Sequence(newName, seqChars); |
| 84 | 33 | updateResidueMap(residueMap, seq); |
| 85 | } else | |
| 86 | { | |
| 87 | // TODO: add ability to exclude hidden regions | |
| 88 | 13 | seq = new Sequence(newName, |
| 89 | AlignSeq.extractGaps(Comparison.GapChars, new String(seqChars))); | |
| 90 | // for annotation need to also record map to sequence start/end | |
| 91 | // position in range | |
| 92 | // then transfer back to original sequence on return. | |
| 93 | } | |
| 94 | 46 | sequences.add(seq); |
| 95 | } | |
| 96 | 24 | boolean[] gapMapArray = null; |
| 97 | 24 | if (submitGaps) |
| 98 | { | |
| 99 | 17 | adjustColumns(sequences, residueMap, requireAligned); |
| 100 | 17 | gapMapArray = new boolean[width]; |
| 101 | 242 | for (int i = 0; i < width; i++) |
| 102 | 225 | gapMapArray[i] = residueMap.get(i); |
| 103 | } | |
| 104 | 24 | return new AnnotationJob(sequences, gapMapArray, namesMap, start, end, |
| 105 | minSize); | |
| 106 | } | |
| 107 | ||
| 108 | 8 | static void replaceNonStandardResidues(char[] seq, char replacement, |
| 109 | boolean isProtein) | |
| 110 | { | |
| 111 | 116 | for (int i = 0; i < seq.length; i++) |
| 112 | { | |
| 113 | 108 | char chr = seq[i]; |
| 114 | 108 | if (isProtein ? ResidueProperties.aaIndex[chr] >= 20 |
| 115 | : ResidueProperties.nucleotideIndex[chr] >= 5) | |
| 116 | { | |
| 117 | 15 | seq[i] = replacement; |
| 118 | } | |
| 119 | } | |
| 120 | } | |
| 121 | ||
| 122 | /** | |
| 123 | * Add residue positions of the given sequence to the residues map. Perform an | |
| 124 | * "or" operation between the given residue map and the inverse of the gap map | |
| 125 | * of the given sequence. | |
| 126 | * | |
| 127 | * @param residueMap | |
| 128 | * mapping to be updated in-place | |
| 129 | * @param seq | |
| 130 | * the sequence whose residue positions are added to the map | |
| 131 | */ | |
| 132 | 33 | static void updateResidueMap(BitSet residueMap, SequenceI seq) |
| 133 | { | |
| 134 | 33 | var gaps = seq.gapBitset(); |
| 135 | 33 | gaps.flip(0, seq.getLength()); |
| 136 | 33 | residueMap.or(gaps); |
| 137 | } | |
| 138 | ||
| 139 | /** | |
| 140 | * Remove columns not included in the mask from the sequences in-place. If | |
| 141 | * {@code padToLength} is set, the shorter sequences are padded with gaps at | |
| 142 | * the end. | |
| 143 | * | |
| 144 | * @param sequences | |
| 145 | * list of sequences to be modified | |
| 146 | * @param mask | |
| 147 | * mask of columns that will remain | |
| 148 | * @param padToLength | |
| 149 | * if gaps should be added to the end of shorter sequences | |
| 150 | */ | |
| 151 | 17 | static void adjustColumns(List<SequenceI> sequences, BitSet mask, |
| 152 | boolean padToLength) | |
| 153 | { | |
| 154 | 17 | int width = mask.cardinality(); |
| 155 | 17 | for (SequenceI seq : sequences) |
| 156 | { | |
| 157 | 33 | char[] chars = SeqsetUtils.filterSequence(seq.getSequence(), mask); |
| 158 | 33 | if (padToLength && chars.length < width) |
| 159 | { | |
| 160 | 2 | int limit = chars.length; |
| 161 | 2 | chars = Arrays.copyOf(chars, width); |
| 162 | 2 | Arrays.fill(chars, limit, chars.length, Comparison.GAP_DASH); |
| 163 | } | |
| 164 | 33 | seq.setEnd(seq.getStart()); |
| 165 | 33 | seq.setSequence(chars); |
| 166 | } | |
| 167 | } | |
| 168 | } |