Coverage Report
File Rna.java
Coverage histogram
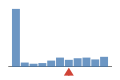
27% of files have more coverage
Classes
| Class | Line # | Actions | |||
|---|---|---|---|---|---|
| Rna | 40 | 147 | 68 |
Contributing tests
This file is covered by 10 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | /* Author: Lauren Michelle Lui | |
| 22 | * Methods are based on RALEE methods http://personalpages.manchester.ac.uk/staff/sam.griffiths-jones/software/ralee/ | |
| 23 | * Additional Author: Jan Engelhart (2011) - Structure consensus and bug fixing | |
| 24 | * Additional Author: Anne Menard (2012) - Pseudoknot support and secondary structure consensus | |
| 25 | * */ | |
| 26 | ||
| 27 | package jalview.analysis; | |
| 28 | ||
| 29 | import jalview.analysis.SecStrConsensus.SimpleBP; | |
| 30 | import jalview.datamodel.SequenceFeature; | |
| 31 | import jalview.util.MessageManager; | |
| 32 | ||
| 33 | import java.util.ArrayList; | |
| 34 | import java.util.HashMap; | |
| 35 | import java.util.Hashtable; | |
| 36 | import java.util.List; | |
| 37 | import java.util.Map; | |
| 38 | import java.util.Stack; | |
| 39 | ||
| 40 | public class Rna | |
| 41 | { | |
| 42 | ||
| 43 | /** | |
| 44 | * Answers true if the character is a valid open pair rna secondary structure | |
| 45 | * symbol. Currently accepts A-Z, ([{< | |
| 46 | * | |
| 47 | * @param c | |
| 48 | * @return | |
| 49 | */ | |
| 50 | 59261 |  public static boolean isOpeningParenthesis(char c) public static boolean isOpeningParenthesis(char c) |
| 51 | { | |
| 52 | 59261 | return ('A' <= c && c <= 'Z' || c == '(' || c == '[' || c == '{' |
| 53 | || c == '<'); | |
| 54 | } | |
| 55 | ||
| 56 | /** | |
| 57 | * Answers true if the string is a valid open pair rna secondary structure | |
| 58 | * symbol. Currently accepts A-Z, ([{< | |
| 59 | * | |
| 60 | * @param s | |
| 61 | * @return | |
| 62 | */ | |
| 63 | 0 | public static boolean isOpeningParenthesis(String s) |
| 64 | { | |
| 65 | 0 | return s != null && s.length() == 1 |
| 66 | && isOpeningParenthesis(s.charAt(0)); | |
| 67 | } | |
| 68 | ||
| 69 | /** | |
| 70 | * Answers true if the character is a valid close pair rna secondary structure | |
| 71 | * symbol. Currently accepts a-z, )]}> | |
| 72 | * | |
| 73 | * @param c | |
| 74 | * @return | |
| 75 | */ | |
| 76 | 43596 | public static boolean isClosingParenthesis(char c) |
| 77 | { | |
| 78 | 43596 | return ('a' <= c && c <= 'z' || c == ')' || c == ']' || c == '}' |
| 79 | || c == '>'); | |
| 80 | } | |
| 81 | ||
| 82 | /** | |
| 83 | * Answers true if the string is a valid close pair rna secondary structure | |
| 84 | * symbol. Currently accepts a-z, )]}> | |
| 85 | * | |
| 86 | * @param s | |
| 87 | * @return | |
| 88 | */ | |
| 89 | 0 | public static boolean isClosingParenthesis(String s) |
| 90 | { | |
| 91 | 0 | return s != null && s.length() == 1 |
| 92 | && isClosingParenthesis(s.charAt(0)); | |
| 93 | } | |
| 94 | ||
| 95 | /** | |
| 96 | * Returns the matching open pair symbol for the given closing symbol. | |
| 97 | * Currently returns A-Z for a-z, or ([{< for )]}>, or the input symbol if it | |
| 98 | * is not a valid closing symbol. | |
| 99 | * | |
| 100 | * @param c | |
| 101 | * @return | |
| 102 | */ | |
| 103 | 9493 | public static char getMatchingOpeningParenthesis(char c) |
| 104 | { | |
| 105 | 9493 | if ('a' <= c && c <= 'z') |
| 106 | { | |
| 107 | 48 | return (char) (c + 'A' - 'a'); |
| 108 | } | |
| 109 | 9445 | switch (c) |
| 110 | { | |
| 111 | 9220 | case ')': |
| 112 | 9220 | return '('; |
| 113 | 68 | case ']': |
| 114 | 68 | return '['; |
| 115 | 36 | case '}': |
| 116 | 36 | return '{'; |
| 117 | 121 | case '>': |
| 118 | 121 | return '<'; |
| 119 | 0 | default: |
| 120 | 0 | return c; |
| 121 | } | |
| 122 | } | |
| 123 | ||
| 124 | /** | |
| 125 | * Based off of RALEE code ralee-get-base-pairs. Keeps track of open bracket | |
| 126 | * positions in "stack" vector. When a close bracket is reached, pair this | |
| 127 | * with the last matching element in the "stack" vector and store in "pairs" | |
| 128 | * vector. Remove last element in the "stack" vector. Continue in this manner | |
| 129 | * until the whole string is processed. Parse errors are thrown as exceptions | |
| 130 | * wrapping the error location - position of the first unmatched closing | |
| 131 | * bracket, or string length if there is an unmatched opening bracket. | |
| 132 | * | |
| 133 | * @param line | |
| 134 | * Secondary structure line of an RNA Stockholm file | |
| 135 | * @return | |
| 136 | * @throw {@link WUSSParseException} | |
| 137 | */ | |
| 138 | 891 | protected static List<SimpleBP> getSimpleBPs(CharSequence line) |
| 139 | throws WUSSParseException | |
| 140 | { | |
| 141 | 891 | Hashtable<Character, Stack<Integer>> stacks = new Hashtable<Character, Stack<Integer>>(); |
| 142 | 891 | List<SimpleBP> pairs = new ArrayList<SimpleBP>(); |
| 143 | 891 | int i = 0; |
| 144 | 53777 | while (i < line.length()) |
| 145 | { | |
| 146 | 52886 | char base = line.charAt(i); |
| 147 | ||
| 148 | 52886 | if (isOpeningParenthesis(base)) |
| 149 | { | |
| 150 | 12217 | if (!stacks.containsKey(base)) |
| 151 | { | |
| 152 | 977 | stacks.put(base, new Stack<Integer>()); |
| 153 | } | |
| 154 | 12217 | stacks.get(base).push(i); |
| 155 | ||
| 156 | } | |
| 157 | 40669 | else if (isClosingParenthesis(base)) |
| 158 | { | |
| 159 | ||
| 160 | 9493 | char opening = getMatchingOpeningParenthesis(base); |
| 161 | ||
| 162 | 9493 | if (!stacks.containsKey(opening)) |
| 163 | { | |
| 164 | 0 | throw new WUSSParseException(MessageManager.formatMessage( |
| 165 | "exception.mismatched_unseen_closing_char", new String[] | |
| 166 | { String.valueOf(base) }), i); | |
| 167 | } | |
| 168 | ||
| 169 | 9493 | Stack<Integer> stack = stacks.get(opening); |
| 170 | 9493 | if (stack.isEmpty()) |
| 171 | { | |
| 172 | // error whilst parsing i'th position. pass back | |
| 173 | 0 | throw new WUSSParseException(MessageManager.formatMessage( |
| 174 | "exception.mismatched_closing_char", new String[] | |
| 175 | { String.valueOf(base) }), i); | |
| 176 | } | |
| 177 | 9493 | int temp = stack.pop(); |
| 178 | ||
| 179 | 9493 | pairs.add(new SimpleBP(temp, i)); |
| 180 | } | |
| 181 | 52886 | i++; |
| 182 | } | |
| 183 | 891 | for (char opening : stacks.keySet()) |
| 184 | { | |
| 185 | 977 | Stack<Integer> stack = stacks.get(opening); |
| 186 | 977 | if (!stack.empty()) |
| 187 | { | |
| 188 | /* | |
| 189 | * we have an unmatched opening bracket; report error as at | |
| 190 | * i (length of input string) | |
| 191 | */ | |
| 192 | 412 | throw new WUSSParseException(MessageManager.formatMessage( |
| 193 | "exception.mismatched_opening_char", new String[] | |
| 194 | { String.valueOf(opening), String.valueOf(stack.pop()) }), | |
| 195 | i); | |
| 196 | } | |
| 197 | } | |
| 198 | 479 | return pairs; |
| 199 | } | |
| 200 | ||
| 201 | /** | |
| 202 | * Function to get the end position corresponding to a given start position | |
| 203 | * | |
| 204 | * @param indice | |
| 205 | * - start position of a base pair | |
| 206 | * @return - end position of a base pair | |
| 207 | */ | |
| 208 | /* | |
| 209 | * makes no sense at the moment :( public int findEnd(int indice){ //TODO: | |
| 210 | * Probably extend this to find the start to a given end? //could be done by | |
| 211 | * putting everything twice to the hash ArrayList<Integer> pair = new | |
| 212 | * ArrayList<Integer>(); return pairHash.get(indice); } | |
| 213 | */ | |
| 214 | ||
| 215 | /** | |
| 216 | * Answers true if the character is a recognised symbol for RNA secondary | |
| 217 | * structure. Currently accepts a-z, A-Z, ()[]{}<>. | |
| 218 | * | |
| 219 | * @param c | |
| 220 | * @return | |
| 221 | */ | |
| 222 | 6299 | public static boolean isRnaSecondaryStructureSymbol(char c) |
| 223 | { | |
| 224 | 6299 | return isOpeningParenthesis(c) || isClosingParenthesis(c); |
| 225 | } | |
| 226 | ||
| 227 | /** | |
| 228 | * Answers true if the string is a recognised symbol for RNA secondary | |
| 229 | * structure. Currently accepts a-z, A-Z, ()[]{}<>. | |
| 230 | * | |
| 231 | * @param s | |
| 232 | * @return | |
| 233 | */ | |
| 234 | 0 | public static boolean isRnaSecondaryStructureSymbol(String s) |
| 235 | { | |
| 236 | 0 | return isOpeningParenthesis(s) || isClosingParenthesis(s); |
| 237 | } | |
| 238 | ||
| 239 | /** | |
| 240 | * Translates a string to RNA secondary structure representation. Returns the | |
| 241 | * string with any non-SS characters changed to spaces. Accepted characters | |
| 242 | * are a-z, A-Z, and (){}[]<> brackets. | |
| 243 | * | |
| 244 | * @param ssString | |
| 245 | * @return | |
| 246 | */ | |
| 247 | 6299 | public static String getRNASecStrucState(String ssString) |
| 248 | { | |
| 249 | 6299 | if (ssString == null) |
| 250 | { | |
| 251 | 0 | return null; |
| 252 | } | |
| 253 | 6299 | StringBuilder result = new StringBuilder(ssString.length()); |
| 254 | 12598 | for (int i = 0; i < ssString.length(); i++) |
| 255 | { | |
| 256 | 6299 | char c = ssString.charAt(i); |
| 257 | 6299 | result.append(isRnaSecondaryStructureSymbol(c) ? c : " "); |
| 258 | } | |
| 259 | 6299 | return result.toString(); |
| 260 | } | |
| 261 | ||
| 262 | /** | |
| 263 | * Answers true if the base-pair is either a Watson-Crick (A:T/U, C:G) or a | |
| 264 | * wobble (G:T/U) pair (either way round), else false | |
| 265 | * | |
| 266 | * @param first | |
| 267 | * @param second | |
| 268 | * @return | |
| 269 | */ | |
| 270 | 0 | public static boolean isCanonicalOrWobblePair(char first, char second) |
| 271 | { | |
| 272 | 0 | if (first > 'Z') |
| 273 | { | |
| 274 | 0 | first -= 32; |
| 275 | } | |
| 276 | 0 | if (second > 'Z') |
| 277 | { | |
| 278 | 0 | second -= 32; |
| 279 | } | |
| 280 | ||
| 281 | 0 | switch (first) |
| 282 | { | |
| 283 | 0 | case 'A': |
| 284 | 0 | switch (second) |
| 285 | { | |
| 286 | 0 | case 'T': |
| 287 | 0 | case 'U': |
| 288 | 0 | return true; |
| 289 | } | |
| 290 | 0 | break; |
| 291 | 0 | case 'C': |
| 292 | 0 | switch (second) |
| 293 | { | |
| 294 | 0 | case 'G': |
| 295 | 0 | return true; |
| 296 | } | |
| 297 | 0 | break; |
| 298 | 0 | case 'T': |
| 299 | 0 | case 'U': |
| 300 | 0 | switch (second) |
| 301 | { | |
| 302 | 0 | case 'A': |
| 303 | 0 | case 'G': |
| 304 | 0 | return true; |
| 305 | } | |
| 306 | 0 | break; |
| 307 | 0 | case 'G': |
| 308 | 0 | switch (second) |
| 309 | { | |
| 310 | 0 | case 'C': |
| 311 | 0 | case 'T': |
| 312 | 0 | case 'U': |
| 313 | 0 | return true; |
| 314 | } | |
| 315 | 0 | break; |
| 316 | } | |
| 317 | 0 | return false; |
| 318 | } | |
| 319 | ||
| 320 | /** | |
| 321 | * Answers true if the base-pair is Watson-Crick - (A:T/U or C:G, either way | |
| 322 | * round), else false | |
| 323 | * | |
| 324 | * @param first | |
| 325 | * @param second | |
| 326 | * @return | |
| 327 | */ | |
| 328 | 0 | public static boolean isCanonicalPair(char first, char second) |
| 329 | { | |
| 330 | ||
| 331 | 0 | if (first > 'Z') |
| 332 | { | |
| 333 | 0 | first -= 32; |
| 334 | } | |
| 335 | 0 | if (second > 'Z') |
| 336 | { | |
| 337 | 0 | second -= 32; |
| 338 | } | |
| 339 | ||
| 340 | 0 | switch (first) |
| 341 | { | |
| 342 | 0 | case 'A': |
| 343 | 0 | switch (second) |
| 344 | { | |
| 345 | 0 | case 'T': |
| 346 | 0 | case 'U': |
| 347 | 0 | return true; |
| 348 | } | |
| 349 | 0 | break; |
| 350 | 0 | case 'G': |
| 351 | 0 | switch (second) |
| 352 | { | |
| 353 | 0 | case 'C': |
| 354 | 0 | return true; |
| 355 | } | |
| 356 | 0 | break; |
| 357 | 0 | case 'C': |
| 358 | 0 | switch (second) |
| 359 | { | |
| 360 | 0 | case 'G': |
| 361 | 0 | return true; |
| 362 | } | |
| 363 | 0 | break; |
| 364 | 0 | case 'T': |
| 365 | 0 | case 'U': |
| 366 | 0 | switch (second) |
| 367 | { | |
| 368 | 0 | case 'A': |
| 369 | 0 | return true; |
| 370 | } | |
| 371 | 0 | break; |
| 372 | } | |
| 373 | 0 | return false; |
| 374 | } | |
| 375 | ||
| 376 | /** | |
| 377 | * Returns the matching close pair symbol for the given opening symbol. | |
| 378 | * Currently returns a-z for A-Z, or )]}> for ([{<, or the input symbol if it | |
| 379 | * is not a valid opening symbol. | |
| 380 | * | |
| 381 | * @param c | |
| 382 | * @return | |
| 383 | */ | |
| 384 | 52 | public static char getMatchingClosingParenthesis(char c) |
| 385 | { | |
| 386 | 52 | if ('A' <= c && c <= 'Z') |
| 387 | { | |
| 388 | 2 | return (char) (c + 'a' - 'A'); |
| 389 | } | |
| 390 | 50 | switch (c) |
| 391 | { | |
| 392 | 13 | case '(': |
| 393 | 13 | return ')'; |
| 394 | 13 | case '[': |
| 395 | 13 | return ']'; |
| 396 | 11 | case '{': |
| 397 | 11 | return '}'; |
| 398 | 13 | case '<': |
| 399 | 13 | return '>'; |
| 400 | 0 | default: |
| 401 | 0 | return c; |
| 402 | } | |
| 403 | } | |
| 404 | ||
| 405 | 891 | public static SequenceFeature[] getHelixMap(CharSequence rnaAnnotation) |
| 406 | throws WUSSParseException | |
| 407 | { | |
| 408 | 891 | List<SequenceFeature> result = new ArrayList<SequenceFeature>(); |
| 409 | ||
| 410 | 891 | int helix = 0; // Number of helices/current helix |
| 411 | 891 | int lastopen = 0; // Position of last open bracket reviewed |
| 412 | 891 | int lastclose = 9999999; // Position of last close bracket reviewed |
| 413 | ||
| 414 | 891 | Map<Integer, Integer> helices = new HashMap<Integer, Integer>(); |
| 415 | // Keep track of helix number for each position | |
| 416 | ||
| 417 | // Go through each base pair and assign positions a helix | |
| 418 | 891 | List<SimpleBP> bps = getSimpleBPs(rnaAnnotation); |
| 419 | 479 | for (SimpleBP basePair : bps) |
| 420 | { | |
| 421 | 9493 | final int open = basePair.getBP5(); |
| 422 | 9493 | final int close = basePair.getBP3(); |
| 423 | ||
| 424 | // jalview.bin.Console.outPrintln("open " + open + " close " + close); | |
| 425 | // jalview.bin.Console.outPrintln("lastclose " + lastclose + " lastopen " | |
| 426 | // + lastopen); | |
| 427 | ||
| 428 | // we're moving from right to left based on closing pair | |
| 429 | /* | |
| 430 | * catch things like <<..>>..<<..>> | | |
| 431 | */ | |
| 432 | 9493 | if (open > lastclose) |
| 433 | { | |
| 434 | 147 | helix++; |
| 435 | } | |
| 436 | ||
| 437 | /* | |
| 438 | * catch things like <<..<<..>>..<<..>>>> | | |
| 439 | */ | |
| 440 | 9493 | int j = bps.size(); |
| 441 | 254029 | while (--j >= 0) |
| 442 | { | |
| 443 | 244612 | int popen = bps.get(j).getBP5(); |
| 444 | ||
| 445 | // jalview.bin.Console.outPrintln("j " + j + " popen " + popen + " | |
| 446 | // lastopen " | |
| 447 | // +lastopen + " open " + open); | |
| 448 | 244612 | if ((popen < lastopen) && (popen > open)) |
| 449 | { | |
| 450 | 76 | if (helices.containsValue(popen) |
| 451 | && ((helices.get(popen)) == helix)) | |
| 452 | { | |
| 453 | 0 | continue; |
| 454 | } | |
| 455 | else | |
| 456 | { | |
| 457 | 76 | helix++; |
| 458 | 76 | break; |
| 459 | } | |
| 460 | } | |
| 461 | } | |
| 462 | ||
| 463 | // Put positions and helix information into the hashtable | |
| 464 | 9493 | helices.put(open, helix); |
| 465 | 9493 | helices.put(close, helix); |
| 466 | ||
| 467 | // Record helix as featuregroup | |
| 468 | 9493 | result.add(new SequenceFeature("RNA helix", "", open, close, |
| 469 | String.valueOf(helix))); | |
| 470 | ||
| 471 | 9493 | lastopen = open; |
| 472 | 9493 | lastclose = close; |
| 473 | } | |
| 474 | ||
| 475 | 479 | return result.toArray(new SequenceFeature[result.size()]); |
| 476 | } | |
| 477 | } |