Coverage Report
File StockholmFile.java
Coverage histogram
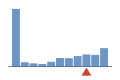
21% of files have more coverage
Classes
| Class | Line # | Actions | |||
|---|---|---|---|---|---|
| StockholmFile | 75 | 481 | 177 |
Contributing tests
This file is covered by 24 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | /* | |
| 22 | * This extension was written by Benjamin Schuster-Boeckler at sanger.ac.uk | |
| 23 | */ | |
| 24 | package jalview.io; | |
| 25 | ||
| 26 | import java.io.BufferedReader; | |
| 27 | import java.io.FileReader; | |
| 28 | import java.io.IOException; | |
| 29 | import java.util.ArrayList; | |
| 30 | import java.util.Enumeration; | |
| 31 | import java.util.Hashtable; | |
| 32 | import java.util.LinkedHashMap; | |
| 33 | import java.util.List; | |
| 34 | import java.util.Locale; | |
| 35 | import java.util.Map; | |
| 36 | import java.util.Vector; | |
| 37 | ||
| 38 | import com.stevesoft.pat.Regex; | |
| 39 | ||
| 40 | import fr.orsay.lri.varna.exceptions.ExceptionUnmatchedClosingParentheses; | |
| 41 | import fr.orsay.lri.varna.factories.RNAFactory; | |
| 42 | import fr.orsay.lri.varna.models.rna.RNA; | |
| 43 | import jalview.analysis.Rna; | |
| 44 | import jalview.datamodel.AlignmentAnnotation; | |
| 45 | import jalview.datamodel.AlignmentI; | |
| 46 | import jalview.datamodel.Annotation; | |
| 47 | import jalview.datamodel.DBRefEntry; | |
| 48 | import jalview.datamodel.DBRefSource; | |
| 49 | import jalview.datamodel.Mapping; | |
| 50 | import jalview.datamodel.Sequence; | |
| 51 | import jalview.datamodel.SequenceFeature; | |
| 52 | import jalview.datamodel.SequenceI; | |
| 53 | import jalview.schemes.ResidueProperties; | |
| 54 | import jalview.util.Comparison; | |
| 55 | import jalview.util.DBRefUtils; | |
| 56 | import jalview.util.Format; | |
| 57 | import jalview.util.MessageManager; | |
| 58 | ||
| 59 | /** | |
| 60 | * This class is supposed to parse a Stockholm format file into Jalview There | |
| 61 | * are TODOs in this class: we do not know what the database source and version | |
| 62 | * is for the file when parsing the #GS= AC tag which associates accessions with | |
| 63 | * sequences. Database references are also not parsed correctly: a separate | |
| 64 | * reference string parser must be added to parse the database reference form | |
| 65 | * into Jalview's local representation. | |
| 66 | * | |
| 67 | * @author bsb at sanger.ac.uk | |
| 68 | * @author Natasha Shersnev (Dundee, UK) (Stockholm file writer) | |
| 69 | * @author Lauren Lui (UCSC, USA) (RNA secondary structure annotation import as | |
| 70 | * stockholm) | |
| 71 | * @author Anne Menard (Paris, FR) (VARNA parsing of Stockholm file data) | |
| 72 | * @version 0.3 + jalview mods | |
| 73 | * | |
| 74 | */ | |
| 75 | public class StockholmFile extends AlignFile | |
| 76 | { | |
| 77 | private static final String ANNOTATION = "annotation"; | |
| 78 | ||
| 79 | // private static final Regex OPEN_PAREN = new Regex("(<|\\[)", "("); | |
| 80 | // | |
| 81 | // private static final Regex CLOSE_PAREN = new Regex("(>|\\])", ")"); | |
| 82 | ||
| 83 | public static final Regex DETECT_BRACKETS = new Regex( | |
| 84 | "(<|>|\\[|\\]|\\(|\\)|\\{|\\})"); | |
| 85 | ||
| 86 | // WUSS extended symbols. Avoid ambiguity with protein SS annotations by using | |
| 87 | // NOT_RNASS first. | |
| 88 | public static final String RNASS_BRACKETS = "<>[](){}AaBbCcDdEeFfGgHhIiJjKkLlMmNnOoPpQqRrSsTtUuVvWwXxYyZz"; | |
| 89 | ||
| 90 | // use the following regex to decide an annotations (whole) line is NOT an RNA | |
| 91 | // SS (it contains only E,H,e,h and other non-brace/non-alpha chars) | |
| 92 | private static final Regex NOT_RNASS = new Regex( | |
| 93 | "^[^<>[\\](){}ADFJ-RUVWYZadfj-ruvwyz]*$"); | |
| 94 | ||
| 95 | StringBuffer out; // output buffer | |
| 96 | ||
| 97 | AlignmentI al; | |
| 98 | ||
| 99 | 0 |  public StockholmFile() public StockholmFile() |
| 100 | { | |
| 101 | } | |
| 102 | ||
| 103 | /** | |
| 104 | * Creates a new StockholmFile object for output. | |
| 105 | */ | |
| 106 | 60 | public StockholmFile(AlignmentI al) |
| 107 | { | |
| 108 | 60 | this.al = al; |
| 109 | } | |
| 110 | ||
| 111 | 0 | public StockholmFile(String inFile, DataSourceType type) |
| 112 | throws IOException | |
| 113 | { | |
| 114 | 0 | super(inFile, type); |
| 115 | } | |
| 116 | ||
| 117 | 45 | public StockholmFile(FileParse source) throws IOException |
| 118 | { | |
| 119 | 45 | super(source); |
| 120 | } | |
| 121 | ||
| 122 | 105 | @Override |
| 123 | public void initData() | |
| 124 | { | |
| 125 | 105 | super.initData(); |
| 126 | } | |
| 127 | ||
| 128 | /** | |
| 129 | * Parse a file in Stockholm format into Jalview's data model using VARNA | |
| 130 | * | |
| 131 | * @throws IOException | |
| 132 | * If there is an error with the input file | |
| 133 | */ | |
| 134 | 0 | public void parse_with_VARNA(java.io.File inFile) throws IOException |
| 135 | { | |
| 136 | 0 | FileReader fr = null; |
| 137 | 0 | fr = new FileReader(inFile); |
| 138 | ||
| 139 | 0 | BufferedReader r = new BufferedReader(fr); |
| 140 | 0 | List<RNA> result = null; |
| 141 | 0 | try |
| 142 | { | |
| 143 | 0 | result = RNAFactory.loadSecStrStockholm(r); |
| 144 | } catch (ExceptionUnmatchedClosingParentheses umcp) | |
| 145 | { | |
| 146 | 0 | errormessage = "Unmatched parentheses in annotation. Aborting (" |
| 147 | + umcp.getMessage() + ")"; | |
| 148 | 0 | throw new IOException(umcp); |
| 149 | } | |
| 150 | // DEBUG jalview.bin.Console.outPrintln("this is the secondary scructure:" | |
| 151 | // +result.size()); | |
| 152 | 0 | SequenceI[] seqs = new SequenceI[result.size()]; |
| 153 | 0 | String id = null; |
| 154 | 0 | for (int i = 0; i < result.size(); i++) |
| 155 | { | |
| 156 | // DEBUG jalview.bin.Console.errPrintln("Processing i'th sequence in | |
| 157 | // Stockholm file") | |
| 158 | 0 | RNA current = result.get(i); |
| 159 | ||
| 160 | 0 | String seq = current.getSeq(); |
| 161 | 0 | String rna = current.getStructDBN(true); |
| 162 | // DEBUG jalview.bin.Console.outPrintln(seq); | |
| 163 | // DEBUG jalview.bin.Console.errPrintln(rna); | |
| 164 | 0 | int begin = 0; |
| 165 | 0 | int end = seq.length() - 1; |
| 166 | 0 | id = safeName(getDataName()); |
| 167 | 0 | seqs[i] = new Sequence(id, seq, begin, end); |
| 168 | 0 | String[] annot = new String[rna.length()]; |
| 169 | 0 | Annotation[] ann = new Annotation[rna.length()]; |
| 170 | 0 | for (int j = 0; j < rna.length(); j++) |
| 171 | { | |
| 172 | 0 | annot[j] = rna.substring(j, j + 1); |
| 173 | ||
| 174 | } | |
| 175 | ||
| 176 | 0 | for (int k = 0; k < rna.length(); k++) |
| 177 | { | |
| 178 | 0 | ann[k] = new Annotation(annot[k], "", |
| 179 | Rna.getRNASecStrucState(annot[k]).charAt(0), 0f); | |
| 180 | ||
| 181 | } | |
| 182 | 0 | AlignmentAnnotation align = new AlignmentAnnotation("Sec. str.", |
| 183 | current.getID(), ann); | |
| 184 | ||
| 185 | 0 | seqs[i].addAlignmentAnnotation(align); |
| 186 | 0 | seqs[i].setRNA(result.get(i)); |
| 187 | 0 | this.annotations.addElement(align); |
| 188 | } | |
| 189 | 0 | this.setSeqs(seqs); |
| 190 | ||
| 191 | } | |
| 192 | ||
| 193 | /** | |
| 194 | * Parse a file in Stockholm format into Jalview's data model. The file has to | |
| 195 | * be passed at construction time | |
| 196 | * | |
| 197 | * @throws IOException | |
| 198 | * If there is an error with the input file | |
| 199 | */ | |
| 200 | 45 | @Override |
| 201 | public void parse() throws IOException | |
| 202 | { | |
| 203 | 45 | StringBuffer treeString = new StringBuffer(); |
| 204 | 45 | String treeName = null; |
| 205 | // --------------- Variable Definitions ------------------- | |
| 206 | 45 | String line; |
| 207 | 45 | String version; |
| 208 | // String id; | |
| 209 | 45 | Hashtable seqAnn = new Hashtable(); // Sequence related annotations |
| 210 | 45 | LinkedHashMap<String, String> seqs = new LinkedHashMap<>(); |
| 211 | 45 | Regex p, r, rend, s, x; |
| 212 | // Temporary line for processing RNA annotation | |
| 213 | // String RNAannot = ""; | |
| 214 | ||
| 215 | // ------------------ Parsing File ---------------------- | |
| 216 | // First, we have to check that this file has STOCKHOLM format, i.e. the | |
| 217 | // first line must match | |
| 218 | ||
| 219 | 45 | r = new Regex("# STOCKHOLM ([\\d\\.]+)"); |
| 220 | 45 | if (!r.search(nextLine())) |
| 221 | { | |
| 222 | 0 | throw new IOException( |
| 223 | MessageManager.getString("exception.stockholm_invalid_format") | |
| 224 | + " (" + r + ")"); | |
| 225 | } | |
| 226 | else | |
| 227 | { | |
| 228 | 45 | version = r.stringMatched(1); |
| 229 | ||
| 230 | // logger.debug("Stockholm version: " + version); | |
| 231 | } | |
| 232 | ||
| 233 | // We define some Regexes here that will be used regularily later | |
| 234 | 45 | rend = new Regex("^\\s*\\/\\/"); // Find the end of an alignment |
| 235 | 45 | p = new Regex("(\\S+)\\/(\\d+)\\-(\\d+)"); // split sequence id in |
| 236 | // id/from/to | |
| 237 | 45 | s = new Regex("(\\S+)\\s+(\\S*)\\s+(.*)"); // Parses annotation subtype |
| 238 | 45 | r = new Regex("#=(G[FSRC]?)\\s+(.*)"); // Finds any annotation line |
| 239 | 45 | x = new Regex("(\\S+)\\s+(\\S+)"); // split id from sequence |
| 240 | ||
| 241 | // Convert all bracket types to parentheses (necessary for passing to VARNA) | |
| 242 | 45 | Regex openparen = new Regex("(<|\\[)", "("); |
| 243 | 45 | Regex closeparen = new Regex("(>|\\])", ")"); |
| 244 | ||
| 245 | // // Detect if file is RNA by looking for bracket types | |
| 246 | // Regex detectbrackets = new Regex("(<|>|\\[|\\]|\\(|\\))"); | |
| 247 | ||
| 248 | 45 | rend.optimize(); |
| 249 | 45 | p.optimize(); |
| 250 | 45 | s.optimize(); |
| 251 | 45 | r.optimize(); |
| 252 | 45 | x.optimize(); |
| 253 | 45 | openparen.optimize(); |
| 254 | 45 | closeparen.optimize(); |
| 255 | ||
| 256 | ? | while ((line = nextLine()) != null) |
| 257 | { | |
| 258 | 2512 | if (line.length() == 0) |
| 259 | { | |
| 260 | 6 | continue; |
| 261 | } | |
| 262 | 2506 | if (rend.search(line)) |
| 263 | { | |
| 264 | // End of the alignment, pass stuff back | |
| 265 | 45 | this.noSeqs = seqs.size(); |
| 266 | ||
| 267 | 45 | String dbsource = null; |
| 268 | 45 | Regex pf = new Regex("PF[0-9]{5}(.*)"); // Finds AC for Pfam |
| 269 | 45 | Regex rf = new Regex("RF[0-9]{5}(.*)"); // Finds AC for Rfam |
| 270 | 45 | if (getAlignmentProperty("AC") != null) |
| 271 | { | |
| 272 | 6 | String dbType = getAlignmentProperty("AC").toString(); |
| 273 | 6 | if (pf.search(dbType)) |
| 274 | { | |
| 275 | // PFAM Alignment - so references are typically from Uniprot | |
| 276 | 3 | dbsource = "PFAM"; |
| 277 | } | |
| 278 | 3 | else if (rf.search(dbType)) |
| 279 | { | |
| 280 | 3 | dbsource = "RFAM"; |
| 281 | } | |
| 282 | } | |
| 283 | // logger.debug("Number of sequences: " + this.noSeqs); | |
| 284 | 45 | for (Map.Entry<String, String> skey : seqs.entrySet()) |
| 285 | { | |
| 286 | // logger.debug("Processing sequence " + acc); | |
| 287 | 917 | String acc = skey.getKey(); |
| 288 | 917 | String seq = skey.getValue(); |
| 289 | 917 | if (maxLength < seq.length()) |
| 290 | { | |
| 291 | 45 | maxLength = seq.length(); |
| 292 | } | |
| 293 | 917 | int start = 1; |
| 294 | 917 | int end = -1; |
| 295 | 917 | String sid = acc; |
| 296 | /* | |
| 297 | * Retrieve hash of annotations for this accession Associate | |
| 298 | * Annotation with accession | |
| 299 | */ | |
| 300 | 917 | Hashtable accAnnotations = null; |
| 301 | ||
| 302 | 917 | if (seqAnn != null && seqAnn.containsKey(acc)) |
| 303 | { | |
| 304 | 916 | accAnnotations = (Hashtable) seqAnn.remove(acc); |
| 305 | // TODO: add structures to sequence | |
| 306 | } | |
| 307 | ||
| 308 | // Split accession in id and from/to | |
| 309 | 917 | if (p.search(acc)) |
| 310 | { | |
| 311 | 684 | sid = p.stringMatched(1); |
| 312 | 684 | start = Integer.parseInt(p.stringMatched(2)); |
| 313 | 684 | end = Integer.parseInt(p.stringMatched(3)); |
| 314 | } | |
| 315 | // logger.debug(sid + ", " + start + ", " + end); | |
| 316 | ||
| 317 | 917 | Sequence seqO = new Sequence(sid, seq, start, end); |
| 318 | // Add Description (if any) | |
| 319 | 917 | if (accAnnotations != null && accAnnotations.containsKey("DE")) |
| 320 | { | |
| 321 | 16 | String desc = (String) accAnnotations.get("DE"); |
| 322 | 16 | seqO.setDescription((desc == null) ? "" : desc); |
| 323 | } | |
| 324 | // Add DB References (if any) | |
| 325 | 917 | if (accAnnotations != null && accAnnotations.containsKey("DR")) |
| 326 | { | |
| 327 | 26 | String dbr = (String) accAnnotations.get("DR"); |
| 328 | 26 | if (dbr != null && dbr.indexOf(";") > -1) |
| 329 | { | |
| 330 | 26 | String src = dbr.substring(0, dbr.indexOf(";")); |
| 331 | 26 | String acn = dbr.substring(dbr.indexOf(";") + 1); |
| 332 | 26 | jalview.util.DBRefUtils.parseToDbRef(seqO, src, "0", acn); |
| 333 | } | |
| 334 | } | |
| 335 | ||
| 336 | 917 | if (accAnnotations != null && accAnnotations.containsKey("AC")) |
| 337 | { | |
| 338 | 895 | String dbr = (String) accAnnotations.get("AC"); |
| 339 | 895 | if (dbr != null) |
| 340 | { | |
| 341 | // we could get very clever here - but for now - just try to | |
| 342 | // guess accession type from type of sequence, source of alignment | |
| 343 | // plus | |
| 344 | // structure | |
| 345 | // of accession | |
| 346 | 895 | guessDatabaseFor(seqO, dbr, dbsource); |
| 347 | } | |
| 348 | // else - do what ? add the data anyway and prompt the user to | |
| 349 | // specify what references these are ? | |
| 350 | } | |
| 351 | ||
| 352 | 917 | Hashtable features = null; |
| 353 | // We need to adjust the positions of all features to account for gaps | |
| 354 | 917 | try |
| 355 | { | |
| 356 | 917 | features = (Hashtable) accAnnotations.remove("features"); |
| 357 | } catch (java.lang.NullPointerException e) | |
| 358 | { | |
| 359 | // loggerwarn("Getting Features for " + acc + ": " + | |
| 360 | // e.getMessage()); | |
| 361 | // continue; | |
| 362 | } | |
| 363 | // if we have features | |
| 364 | 917 | if (features != null) |
| 365 | { | |
| 366 | 320 | int posmap[] = seqO.findPositionMap(); |
| 367 | 320 | Enumeration i = features.keys(); |
| 368 | 640 | while (i.hasMoreElements()) |
| 369 | { | |
| 370 | // TODO: parse out secondary structure annotation as annotation | |
| 371 | // row | |
| 372 | // TODO: parse out scores as annotation row | |
| 373 | // TODO: map coding region to core jalview feature types | |
| 374 | 320 | String type = i.nextElement().toString(); |
| 375 | 320 | Hashtable content = (Hashtable) features.remove(type); |
| 376 | ||
| 377 | // add alignment annotation for this feature | |
| 378 | 320 | String key = type2id(type); |
| 379 | ||
| 380 | /* | |
| 381 | * have we added annotation rows for this type ? | |
| 382 | */ | |
| 383 | 320 | boolean annotsAdded = false; |
| 384 | 320 | if (key != null) |
| 385 | { | |
| 386 | 320 | if (accAnnotations != null |
| 387 | && accAnnotations.containsKey(key)) | |
| 388 | { | |
| 389 | 320 | Vector vv = (Vector) accAnnotations.get(key); |
| 390 | 640 | for (int ii = 0; ii < vv.size(); ii++) |
| 391 | { | |
| 392 | 320 | annotsAdded = true; |
| 393 | 320 | AlignmentAnnotation an = (AlignmentAnnotation) vv |
| 394 | .elementAt(ii); | |
| 395 | 320 | seqO.addAlignmentAnnotation(an); |
| 396 | 320 | annotations.add(an); |
| 397 | } | |
| 398 | } | |
| 399 | } | |
| 400 | ||
| 401 | 320 | Enumeration j = content.keys(); |
| 402 | 640 | while (j.hasMoreElements()) |
| 403 | { | |
| 404 | 320 | String desc = j.nextElement().toString(); |
| 405 | 320 | if (ANNOTATION.equals(desc) && annotsAdded) |
| 406 | { | |
| 407 | // don't add features if we already added an annotation row | |
| 408 | 320 | continue; |
| 409 | } | |
| 410 | 0 | String ns = content.get(desc).toString(); |
| 411 | 0 | char[] byChar = ns.toCharArray(); |
| 412 | 0 | for (int k = 0; k < byChar.length; k++) |
| 413 | { | |
| 414 | 0 | char c = byChar[k]; |
| 415 | 0 | if (!(c == ' ' || c == '_' || c == '-' || c == '.')) // PFAM |
| 416 | // uses | |
| 417 | // '.' | |
| 418 | // for | |
| 419 | // feature | |
| 420 | // background | |
| 421 | { | |
| 422 | 0 | int new_pos = posmap[k]; // look up nearest seqeunce |
| 423 | // position to this column | |
| 424 | 0 | SequenceFeature feat = new SequenceFeature(type, desc, |
| 425 | new_pos, new_pos, null); | |
| 426 | ||
| 427 | 0 | seqO.addSequenceFeature(feat); |
| 428 | } | |
| 429 | } | |
| 430 | } | |
| 431 | ||
| 432 | } | |
| 433 | ||
| 434 | } | |
| 435 | // garbage collect | |
| 436 | ||
| 437 | // logger.debug("Adding seq " + acc + " from " + start + " to " + end | |
| 438 | // + ": " + seq); | |
| 439 | 917 | this.seqs.addElement(seqO); |
| 440 | } | |
| 441 | 45 | return; // finished parsing this segment of source |
| 442 | } | |
| 443 | 2461 | else if (!r.search(line)) |
| 444 | { | |
| 445 | // jalview.bin.Console.errPrintln("Found sequence line: " + line); | |
| 446 | ||
| 447 | // Split sequence in sequence and accession parts | |
| 448 | 917 | if (!x.search(line)) |
| 449 | { | |
| 450 | // logger.error("Could not parse sequence line: " + line); | |
| 451 | 0 | throw new IOException(MessageManager.formatMessage( |
| 452 | "exception.couldnt_parse_sequence_line", new String[] | |
| 453 | { line })); | |
| 454 | } | |
| 455 | 917 | String ns = seqs.get(x.stringMatched(1)); |
| 456 | 917 | if (ns == null) |
| 457 | { | |
| 458 | 917 | ns = ""; |
| 459 | } | |
| 460 | 917 | ns += x.stringMatched(2); |
| 461 | ||
| 462 | 917 | seqs.put(x.stringMatched(1), ns); |
| 463 | } | |
| 464 | else | |
| 465 | { | |
| 466 | 1544 | String annType = r.stringMatched(1); |
| 467 | 1544 | String annContent = r.stringMatched(2); |
| 468 | ||
| 469 | // jalview.bin.Console.errPrintln("type:" + annType + " content: " + | |
| 470 | // annContent); | |
| 471 | ||
| 472 | 1544 | if (annType.equals("GF")) |
| 473 | { | |
| 474 | /* | |
| 475 | * Generic per-File annotation, free text Magic features: #=GF NH | |
| 476 | * <tree in New Hampshire eXtended format> #=GF TN <Unique identifier | |
| 477 | * for the next tree> Pfam descriptions: 7. DESCRIPTION OF FIELDS | |
| 478 | * | |
| 479 | * Compulsory fields: ------------------ | |
| 480 | * | |
| 481 | * AC Accession number: Accession number in form PFxxxxx.version or | |
| 482 | * PBxxxxxx. ID Identification: One word name for family. DE | |
| 483 | * Definition: Short description of family. AU Author: Authors of the | |
| 484 | * entry. SE Source of seed: The source suggesting the seed members | |
| 485 | * belong to one family. GA Gathering method: Search threshold to | |
| 486 | * build the full alignment. TC Trusted Cutoff: Lowest sequence score | |
| 487 | * and domain score of match in the full alignment. NC Noise Cutoff: | |
| 488 | * Highest sequence score and domain score of match not in full | |
| 489 | * alignment. TP Type: Type of family -- presently Family, Domain, | |
| 490 | * Motif or Repeat. SQ Sequence: Number of sequences in alignment. AM | |
| 491 | * Alignment Method The order ls and fs hits are aligned to the model | |
| 492 | * to build the full align. // End of alignment. | |
| 493 | * | |
| 494 | * Optional fields: ---------------- | |
| 495 | * | |
| 496 | * DC Database Comment: Comment about database reference. DR Database | |
| 497 | * Reference: Reference to external database. RC Reference Comment: | |
| 498 | * Comment about literature reference. RN Reference Number: Reference | |
| 499 | * Number. RM Reference Medline: Eight digit medline UI number. RT | |
| 500 | * Reference Title: Reference Title. RA Reference Author: Reference | |
| 501 | * Author RL Reference Location: Journal location. PI Previous | |
| 502 | * identifier: Record of all previous ID lines. KW Keywords: Keywords. | |
| 503 | * CC Comment: Comments. NE Pfam accession: Indicates a nested domain. | |
| 504 | * NL Location: Location of nested domains - sequence ID, start and | |
| 505 | * end of insert. | |
| 506 | * | |
| 507 | * Obsolete fields: ----------- AL Alignment method of seed: The | |
| 508 | * method used to align the seed members. | |
| 509 | */ | |
| 510 | // Let's save the annotations, maybe we'll be able to do something | |
| 511 | // with them later... | |
| 512 | 152 | Regex an = new Regex("(\\w+)\\s*(.*)"); |
| 513 | 152 | if (an.search(annContent)) |
| 514 | { | |
| 515 | 152 | if (an.stringMatched(1).equals("NH")) |
| 516 | { | |
| 517 | 0 | treeString.append(an.stringMatched(2)); |
| 518 | } | |
| 519 | 152 | else if (an.stringMatched(1).equals("TN")) |
| 520 | { | |
| 521 | 0 | if (treeString.length() > 0) |
| 522 | { | |
| 523 | 0 | if (treeName == null) |
| 524 | { | |
| 525 | 0 | treeName = "Tree " + (getTreeCount() + 1); |
| 526 | } | |
| 527 | 0 | addNewickTree(treeName, treeString.toString()); |
| 528 | } | |
| 529 | 0 | treeName = an.stringMatched(2); |
| 530 | 0 | treeString = new StringBuffer(); |
| 531 | } | |
| 532 | // TODO: JAL-3532 - this is where GF comments and database | |
| 533 | // references are lost | |
| 534 | // suggest overriding this method for Stockholm files to catch and | |
| 535 | // properly | |
| 536 | // process CC, DR etc into multivalued properties | |
| 537 | 152 | setAlignmentProperty(an.stringMatched(1), an.stringMatched(2)); |
| 538 | } | |
| 539 | } | |
| 540 | 1392 | else if (annType.equals("GS")) |
| 541 | { | |
| 542 | // Generic per-Sequence annotation, free text | |
| 543 | /* | |
| 544 | * Pfam uses these features: Feature Description --------------------- | |
| 545 | * ----------- AC <accession> ACcession number DE <freetext> | |
| 546 | * DEscription DR <db>; <accession>; Database Reference OS <organism> | |
| 547 | * OrganiSm (species) OC <clade> Organism Classification (clade, etc.) | |
| 548 | * LO <look> Look (Color, etc.) | |
| 549 | */ | |
| 550 | 1059 | if (s.search(annContent)) |
| 551 | { | |
| 552 | 1059 | String acc = s.stringMatched(1); |
| 553 | 1059 | String type = s.stringMatched(2); |
| 554 | 1059 | String content = s.stringMatched(3); |
| 555 | // TODO: store DR in a vector. | |
| 556 | // TODO: store AC according to generic file db annotation. | |
| 557 | 1059 | Hashtable ann; |
| 558 | 1059 | if (seqAnn.containsKey(acc)) |
| 559 | { | |
| 560 | 148 | ann = (Hashtable) seqAnn.get(acc); |
| 561 | } | |
| 562 | else | |
| 563 | { | |
| 564 | 911 | ann = new Hashtable(); |
| 565 | } | |
| 566 | 1059 | ann.put(type, content); |
| 567 | 1059 | seqAnn.put(acc, ann); |
| 568 | } | |
| 569 | else | |
| 570 | { | |
| 571 | // throw new IOException("Error parsing " + line); | |
| 572 | 0 | jalview.bin.Console |
| 573 | .errPrintln(">> missing annotation: " + line); | |
| 574 | } | |
| 575 | } | |
| 576 | 333 | else if (annType.equals("GC")) |
| 577 | { | |
| 578 | // Generic per-Column annotation, exactly 1 char per column | |
| 579 | // always need a label. | |
| 580 | 13 | if (x.search(annContent)) |
| 581 | { | |
| 582 | // parse out and create alignment annotation directly. | |
| 583 | 13 | parseAnnotationRow(annotations, x.stringMatched(1), |
| 584 | x.stringMatched(2)); | |
| 585 | } | |
| 586 | } | |
| 587 | 320 | else if (annType.equals("GR")) |
| 588 | { | |
| 589 | // Generic per-Sequence AND per-Column markup, exactly 1 char per | |
| 590 | // column | |
| 591 | /* | |
| 592 | * Feature Description Markup letters ------- ----------- | |
| 593 | * -------------- SS Secondary Structure [HGIEBTSCX] SA Surface | |
| 594 | * Accessibility [0-9X] (0=0%-10%; ...; 9=90%-100%) TM TransMembrane | |
| 595 | * [Mio] PP Posterior Probability [0-9*] (0=0.00-0.05; 1=0.05-0.15; | |
| 596 | * *=0.95-1.00) LI LIgand binding [*] AS Active Site [*] IN INtron (in | |
| 597 | * or after) [0-2] | |
| 598 | */ | |
| 599 | 320 | if (s.search(annContent)) |
| 600 | { | |
| 601 | 320 | String acc = s.stringMatched(1); |
| 602 | 320 | String type = s.stringMatched(2); |
| 603 | 320 | String oseq = s.stringMatched(3); |
| 604 | /* | |
| 605 | * copy of annotation field that may be processed into whitespace chunks | |
| 606 | */ | |
| 607 | 320 | String seq = new String(oseq); |
| 608 | ||
| 609 | 320 | Hashtable ann; |
| 610 | // Get an object with all the annotations for this sequence | |
| 611 | 320 | if (seqAnn.containsKey(acc)) |
| 612 | { | |
| 613 | // logger.debug("Found annotations for " + acc); | |
| 614 | 315 | ann = (Hashtable) seqAnn.get(acc); |
| 615 | } | |
| 616 | else | |
| 617 | { | |
| 618 | // logger.debug("Creating new annotations holder for " + acc); | |
| 619 | 5 | ann = new Hashtable(); |
| 620 | 5 | seqAnn.put(acc, ann); |
| 621 | } | |
| 622 | ||
| 623 | // // start of block for appending annotation lines for wrapped | |
| 624 | // stokchholm file | |
| 625 | // TODO test structure, call parseAnnotationRow with vector from | |
| 626 | // hashtable for specific sequence | |
| 627 | ||
| 628 | 320 | Hashtable features; |
| 629 | // Get an object with all the content for an annotation | |
| 630 | 320 | if (ann.containsKey("features")) |
| 631 | { | |
| 632 | // logger.debug("Found features for " + acc); | |
| 633 | 0 | features = (Hashtable) ann.get("features"); |
| 634 | } | |
| 635 | else | |
| 636 | { | |
| 637 | // logger.debug("Creating new features holder for " + acc); | |
| 638 | 320 | features = new Hashtable(); |
| 639 | 320 | ann.put("features", features); |
| 640 | } | |
| 641 | ||
| 642 | 320 | Hashtable content; |
| 643 | 320 | if (features.containsKey(this.id2type(type))) |
| 644 | { | |
| 645 | // logger.debug("Found content for " + this.id2type(type)); | |
| 646 | 0 | content = (Hashtable) features.get(this.id2type(type)); |
| 647 | } | |
| 648 | else | |
| 649 | { | |
| 650 | // logger.debug("Creating new content holder for " + | |
| 651 | // this.id2type(type)); | |
| 652 | 320 | content = new Hashtable(); |
| 653 | 320 | features.put(this.id2type(type), content); |
| 654 | } | |
| 655 | 320 | String ns = (String) content.get(ANNOTATION); |
| 656 | ||
| 657 | 320 | if (ns == null) |
| 658 | { | |
| 659 | 320 | ns = ""; |
| 660 | } | |
| 661 | // finally, append the annotation line | |
| 662 | 320 | ns += seq; |
| 663 | 320 | content.put(ANNOTATION, ns); |
| 664 | // // end of wrapped annotation block. | |
| 665 | // // Now a new row is created with the current set of data | |
| 666 | ||
| 667 | 320 | Hashtable strucAnn; |
| 668 | 320 | if (seqAnn.containsKey(acc)) |
| 669 | { | |
| 670 | 320 | strucAnn = (Hashtable) seqAnn.get(acc); |
| 671 | } | |
| 672 | else | |
| 673 | { | |
| 674 | 0 | strucAnn = new Hashtable(); |
| 675 | } | |
| 676 | ||
| 677 | 320 | Vector<AlignmentAnnotation> newStruc = new Vector<>(); |
| 678 | 320 | parseAnnotationRow(newStruc, type, ns); |
| 679 | 320 | for (AlignmentAnnotation alan : newStruc) |
| 680 | { | |
| 681 | 320 | alan.visible = false; |
| 682 | } | |
| 683 | // new annotation overwrites any existing annotation... | |
| 684 | ||
| 685 | 320 | strucAnn.put(type, newStruc); |
| 686 | 320 | seqAnn.put(acc, strucAnn); |
| 687 | } | |
| 688 | // } | |
| 689 | else | |
| 690 | { | |
| 691 | 0 | jalview.bin.Console.errPrintln( |
| 692 | "Warning - couldn't parse sequence annotation row line:\n" | |
| 693 | + line); | |
| 694 | // throw new IOException("Error parsing " + line); | |
| 695 | } | |
| 696 | } | |
| 697 | else | |
| 698 | { | |
| 699 | 0 | throw new IOException(MessageManager.formatMessage( |
| 700 | "exception.unknown_annotation_detected", new String[] | |
| 701 | { annType, annContent })); | |
| 702 | } | |
| 703 | } | |
| 704 | } | |
| 705 | 0 | if (treeString.length() > 0) |
| 706 | { | |
| 707 | 0 | if (treeName == null) |
| 708 | { | |
| 709 | 0 | treeName = "Tree " + (1 + getTreeCount()); |
| 710 | } | |
| 711 | 0 | addNewickTree(treeName, treeString.toString()); |
| 712 | } | |
| 713 | } | |
| 714 | ||
| 715 | /** | |
| 716 | * Demangle an accession string and guess the originating sequence database | |
| 717 | * for a given sequence | |
| 718 | * | |
| 719 | * @param seqO | |
| 720 | * sequence to be annotated | |
| 721 | * @param dbr | |
| 722 | * Accession string for sequence | |
| 723 | * @param dbsource | |
| 724 | * source database for alignment (PFAM or RFAM) | |
| 725 | */ | |
| 726 | 895 | private void guessDatabaseFor(Sequence seqO, String dbr, String dbsource) |
| 727 | { | |
| 728 | 895 | DBRefEntry dbrf = null; |
| 729 | 895 | List<DBRefEntry> dbrs = new ArrayList<>(); |
| 730 | 895 | String seqdb = "Unknown", sdbac = "" + dbr; |
| 731 | 895 | int st = -1, en = -1, p; |
| 732 | ? | if ((st = sdbac.indexOf("/")) > -1) |
| 733 | { | |
| 734 | 215 | String num, range = sdbac.substring(st + 1); |
| 735 | 215 | sdbac = sdbac.substring(0, st); |
| 736 | ? | if ((p = range.indexOf("-")) > -1) |
| 737 | { | |
| 738 | 215 | p++; |
| 739 | 215 | if (p < range.length()) |
| 740 | { | |
| 741 | 215 | num = range.substring(p).trim(); |
| 742 | 215 | try |
| 743 | { | |
| 744 | 215 | en = Integer.parseInt(num); |
| 745 | } catch (NumberFormatException x) | |
| 746 | { | |
| 747 | // could warn here that index is invalid | |
| 748 | 0 | en = -1; |
| 749 | } | |
| 750 | } | |
| 751 | } | |
| 752 | else | |
| 753 | { | |
| 754 | 0 | p = range.length(); |
| 755 | } | |
| 756 | 215 | num = range.substring(0, p).trim(); |
| 757 | 215 | try |
| 758 | { | |
| 759 | 215 | st = Integer.parseInt(num); |
| 760 | } catch (NumberFormatException x) | |
| 761 | { | |
| 762 | // could warn here that index is invalid | |
| 763 | 215 | st = -1; |
| 764 | } | |
| 765 | } | |
| 766 | 895 | if (dbsource == null) |
| 767 | { | |
| 768 | // make up an origin based on whether the sequence looks like it is | |
| 769 | // nucleotide | |
| 770 | // or protein | |
| 771 | 94 | dbsource = (seqO.isProtein()) ? "PFAM" : "RFAM"; |
| 772 | } | |
| 773 | 895 | if (dbsource.equals("PFAM")) |
| 774 | { | |
| 775 | 619 | seqdb = "UNIPROT"; |
| 776 | 619 | if (sdbac.indexOf(".") > -1) |
| 777 | { | |
| 778 | // strip of last subdomain | |
| 779 | 412 | sdbac = sdbac.substring(0, sdbac.indexOf(".")); |
| 780 | 412 | dbrf = jalview.util.DBRefUtils.parseToDbRef(seqO, seqdb, dbsource, |
| 781 | sdbac); | |
| 782 | 412 | if (dbrf != null) |
| 783 | { | |
| 784 | 412 | dbrs.add(dbrf); |
| 785 | } | |
| 786 | } | |
| 787 | 619 | dbrf = jalview.util.DBRefUtils.parseToDbRef(seqO, dbsource, dbsource, |
| 788 | dbr); | |
| 789 | 619 | if (dbr != null) |
| 790 | { | |
| 791 | 619 | dbrs.add(dbrf); |
| 792 | } | |
| 793 | } | |
| 794 | else | |
| 795 | { | |
| 796 | 276 | seqdb = "EMBL"; // total guess - could be ENA, or something else these |
| 797 | // days | |
| 798 | 276 | if (sdbac.indexOf(".") > -1) |
| 799 | { | |
| 800 | // strip off last subdomain | |
| 801 | 215 | sdbac = sdbac.substring(0, sdbac.indexOf(".")); |
| 802 | 215 | dbrf = jalview.util.DBRefUtils.parseToDbRef(seqO, seqdb, dbsource, |
| 803 | sdbac); | |
| 804 | 215 | if (dbrf != null) |
| 805 | { | |
| 806 | 215 | dbrs.add(dbrf); |
| 807 | } | |
| 808 | } | |
| 809 | ||
| 810 | 276 | dbrf = jalview.util.DBRefUtils.parseToDbRef(seqO, dbsource, dbsource, |
| 811 | dbr); | |
| 812 | 276 | if (dbrf != null) |
| 813 | { | |
| 814 | 276 | dbrs.add(dbrf); |
| 815 | } | |
| 816 | } | |
| 817 | 895 | if (st != -1 && en != -1) |
| 818 | { | |
| 819 | 0 | for (DBRefEntry d : dbrs) |
| 820 | { | |
| 821 | 0 | jalview.util.MapList mp = new jalview.util.MapList( |
| 822 | new int[] | |
| 823 | { seqO.getStart(), seqO.getEnd() }, new int[] { st, en }, 1, | |
| 824 | 1); | |
| 825 | 0 | jalview.datamodel.Mapping mping = new Mapping(mp); |
| 826 | 0 | d.setMap(mping); |
| 827 | } | |
| 828 | } | |
| 829 | } | |
| 830 | ||
| 831 | 333 | protected static AlignmentAnnotation parseAnnotationRow( |
| 832 | Vector<AlignmentAnnotation> annotation, String label, | |
| 833 | String annots) | |
| 834 | { | |
| 835 | 333 | String convert1, convert2 = null; |
| 836 | ||
| 837 | // convert1 = OPEN_PAREN.replaceAll(annots); | |
| 838 | // convert2 = CLOSE_PAREN.replaceAll(convert1); | |
| 839 | // annots = convert2; | |
| 840 | ||
| 841 | 333 | String type = label; |
| 842 | 333 | if (label.contains("_cons")) |
| 843 | { | |
| 844 | 11 | type = (label.indexOf("_cons") == label.length() - 5) |
| 845 | ? label.substring(0, label.length() - 5) | |
| 846 | : label; | |
| 847 | } | |
| 848 | 333 | boolean ss = false, posterior = false; |
| 849 | 333 | type = id2type(type); |
| 850 | ||
| 851 | 333 | boolean isrnass = false; |
| 852 | 333 | if (type.equalsIgnoreCase("secondary structure")) |
| 853 | { | |
| 854 | 327 | ss = true; |
| 855 | 327 | isrnass = !NOT_RNASS.search(annots); // sorry about the double negative |
| 856 | // here (it's easier for dealing with | |
| 857 | // other non-alpha-non-brace chars) | |
| 858 | } | |
| 859 | 333 | if (type.equalsIgnoreCase("posterior probability")) |
| 860 | { | |
| 861 | 0 | posterior = true; |
| 862 | } | |
| 863 | // decide on secondary structure or not. | |
| 864 | 333 | Annotation[] els = new Annotation[annots.length()]; |
| 865 | 25982 | for (int i = 0; i < annots.length(); i++) |
| 866 | { | |
| 867 | 25649 | String pos = annots.substring(i, i + 1); |
| 868 | 25649 | Annotation ann; |
| 869 | 25649 | ann = new Annotation(pos, "", ' ', 0f); // 0f is 'valid' null - will not |
| 870 | // be written out | |
| 871 | 25649 | if (ss) |
| 872 | { | |
| 873 | // if (" .-_".indexOf(pos) == -1) | |
| 874 | { | |
| 875 | 24998 | if (isrnass && RNASS_BRACKETS.indexOf(pos) >= 0) |
| 876 | { | |
| 877 | 8020 | ann.secondaryStructure = Rna.getRNASecStrucState(pos).charAt(0); |
| 878 | 8020 | ann.displayCharacter = "" + pos.charAt(0); |
| 879 | } | |
| 880 | else | |
| 881 | { | |
| 882 | 16978 | ann.secondaryStructure = ResidueProperties.getDssp3state(pos) |
| 883 | .charAt(0); | |
| 884 | ||
| 885 | 16978 | if (ann.secondaryStructure == pos.charAt(0)) |
| 886 | { | |
| 887 | 1766 | ann.displayCharacter = ""; // null; // " "; |
| 888 | } | |
| 889 | else | |
| 890 | { | |
| 891 | 15212 | ann.displayCharacter = " " + ann.displayCharacter; |
| 892 | } | |
| 893 | } | |
| 894 | } | |
| 895 | ||
| 896 | } | |
| 897 | 25649 | if (posterior && !ann.isWhitespace() |
| 898 | && !Comparison.isGap(pos.charAt(0))) | |
| 899 | { | |
| 900 | 0 | float val = 0; |
| 901 | // symbol encodes values - 0..*==0..10 | |
| 902 | 0 | if (pos.charAt(0) == '*') |
| 903 | { | |
| 904 | 0 | val = 10; |
| 905 | } | |
| 906 | else | |
| 907 | { | |
| 908 | 0 | val = pos.charAt(0) - '0'; |
| 909 | 0 | if (val > 9) |
| 910 | { | |
| 911 | 0 | val = 10; |
| 912 | } | |
| 913 | } | |
| 914 | 0 | ann.value = val; |
| 915 | } | |
| 916 | ||
| 917 | 25649 | els[i] = ann; |
| 918 | } | |
| 919 | 333 | AlignmentAnnotation annot = null; |
| 920 | 333 | Enumeration<AlignmentAnnotation> e = annotation.elements(); |
| 921 | 339 | while (e.hasMoreElements()) |
| 922 | { | |
| 923 | 6 | annot = e.nextElement(); |
| 924 | 6 | if (annot.label.equals(type)) |
| 925 | { | |
| 926 | 0 | break; |
| 927 | } | |
| 928 | 6 | annot = null; |
| 929 | } | |
| 930 | 333 | if (annot == null) |
| 931 | { | |
| 932 | 333 | annot = new AlignmentAnnotation(type, type, els); |
| 933 | 333 | annotation.addElement(annot); |
| 934 | } | |
| 935 | else | |
| 936 | { | |
| 937 | 0 | Annotation[] anns = new Annotation[annot.annotations.length |
| 938 | + els.length]; | |
| 939 | 0 | System.arraycopy(annot.annotations, 0, anns, 0, |
| 940 | annot.annotations.length); | |
| 941 | 0 | System.arraycopy(els, 0, anns, annot.annotations.length, els.length); |
| 942 | 0 | annot.annotations = anns; |
| 943 | // jalview.bin.Console.outPrintln("else: "); | |
| 944 | } | |
| 945 | 333 | return annot; |
| 946 | } | |
| 947 | ||
| 948 | 281 | private String dbref_to_ac_record(DBRefEntry ref) |
| 949 | { | |
| 950 | 281 | return ref.getSource().toString() + " ; " |
| 951 | + ref.getAccessionId().toString(); | |
| 952 | } | |
| 953 | ||
| 954 | 60 | @Override |
| 955 | public String print(SequenceI[] s, boolean jvSuffix) | |
| 956 | { | |
| 957 | 60 | out = new StringBuffer(); |
| 958 | 60 | out.append("# STOCKHOLM 1.0"); |
| 959 | 60 | out.append(newline); |
| 960 | ||
| 961 | // find max length of id | |
| 962 | 60 | int max = 0; |
| 963 | 60 | int maxid = 0; |
| 964 | 60 | int in = 0; |
| 965 | 60 | int slen = s.length; |
| 966 | 60 | SequenceI seq; |
| 967 | 60 | Hashtable<String, String> dataRef = null; |
| 968 | 60 | boolean isAA = s[in].isProtein(); |
| 969 | ? | while ((in < slen) && ((seq = s[in]) != null)) |
| 970 | { | |
| 971 | 344 | String tmp = printId(seq, jvSuffix); |
| 972 | 344 | max = Math.max(max, seq.getLength()); |
| 973 | ||
| 974 | 344 | if (tmp.length() > maxid) |
| 975 | { | |
| 976 | 65 | maxid = tmp.length(); |
| 977 | } | |
| 978 | 344 | List<DBRefEntry> seqrefs = seq.getDBRefs(); |
| 979 | 344 | int ndb; |
| 980 | ? | if (seqrefs != null && (ndb = seqrefs.size()) > 0) |
| 981 | { | |
| 982 | 268 | if (dataRef == null) |
| 983 | { | |
| 984 | 3 | dataRef = new Hashtable<>(); |
| 985 | } | |
| 986 | 268 | List<DBRefEntry> primrefs = seq.getPrimaryDBRefs(); |
| 987 | 268 | if (primrefs.size() >= 1) |
| 988 | { | |
| 989 | 1 | dataRef.put(tmp, dbref_to_ac_record(primrefs.get(0))); |
| 990 | } | |
| 991 | else | |
| 992 | { | |
| 993 | 280 | for (int idb = 0; idb < seq.getDBRefs().size(); idb++) |
| 994 | { | |
| 995 | 280 | DBRefEntry dbref = seq.getDBRefs().get(idb); |
| 996 | 280 | dataRef.put(tmp, dbref_to_ac_record(dbref)); |
| 997 | // if we put in a uniprot or EMBL record then we're done: | |
| 998 | 280 | if (isAA && DBRefSource.UNIPROT |
| 999 | .equals(DBRefUtils.getCanonicalName(dbref.getSource()))) | |
| 1000 | { | |
| 1001 | 206 | break; |
| 1002 | } | |
| 1003 | 74 | if (!isAA && DBRefSource.EMBL |
| 1004 | .equals(DBRefUtils.getCanonicalName(dbref.getSource()))) | |
| 1005 | { | |
| 1006 | 61 | break; |
| 1007 | } | |
| 1008 | } | |
| 1009 | } | |
| 1010 | } | |
| 1011 | 344 | in++; |
| 1012 | } | |
| 1013 | 60 | maxid += 9; |
| 1014 | 60 | int i = 0; |
| 1015 | ||
| 1016 | // output database type | |
| 1017 | 60 | if (al.getProperties() != null) |
| 1018 | { | |
| 1019 | 3 | if (!al.getProperties().isEmpty()) |
| 1020 | { | |
| 1021 | 3 | Enumeration key = al.getProperties().keys(); |
| 1022 | 3 | Enumeration val = al.getProperties().elements(); |
| 1023 | 41 | while (key.hasMoreElements()) |
| 1024 | { | |
| 1025 | 38 | out.append("#=GF " + key.nextElement() + " " + val.nextElement()); |
| 1026 | 38 | out.append(newline); |
| 1027 | } | |
| 1028 | } | |
| 1029 | } | |
| 1030 | ||
| 1031 | // output database accessions | |
| 1032 | 60 | if (dataRef != null) |
| 1033 | { | |
| 1034 | 3 | Enumeration<String> en = dataRef.keys(); |
| 1035 | 271 | while (en.hasMoreElements()) |
| 1036 | { | |
| 1037 | 268 | Object idd = en.nextElement(); |
| 1038 | 268 | String type = dataRef.remove(idd); |
| 1039 | 268 | out.append(new Format("%-" + (maxid - 2) + "s") |
| 1040 | .form("#=GS " + idd.toString() + " ")); | |
| 1041 | 268 | if (isAA && type.contains("UNIPROT") |
| 1042 | || (!isAA && type.contains("EMBL"))) | |
| 1043 | { | |
| 1044 | ||
| 1045 | 268 | out.append(" AC " + type.substring(type.indexOf(";") + 1)); |
| 1046 | } | |
| 1047 | else | |
| 1048 | { | |
| 1049 | 0 | out.append(" DR " + type + " "); |
| 1050 | } | |
| 1051 | 268 | out.append(newline); |
| 1052 | } | |
| 1053 | } | |
| 1054 | ||
| 1055 | // output description and annotations | |
| 1056 | ||
| 1057 | ? | while (i < slen && (seq = s[i]) != null) |
| 1058 | { | |
| 1059 | 344 | if (seq.getDescription() != null) |
| 1060 | { | |
| 1061 | // out.append("#=GR "); | |
| 1062 | 16 | out.append(new Format("%-" + maxid + "s").form("#=GS " |
| 1063 | + printId(seq, jvSuffix) + " DE " + seq.getDescription())); | |
| 1064 | 16 | out.append(newline); |
| 1065 | } | |
| 1066 | ||
| 1067 | 344 | AlignmentAnnotation[] alAnot = seq.getAnnotation(); |
| 1068 | 344 | if (alAnot != null) |
| 1069 | { | |
| 1070 | 83 | Annotation[] ann; |
| 1071 | 166 | for (int j = 0, nj = alAnot.length; j < nj; j++) |
| 1072 | { | |
| 1073 | ||
| 1074 | 83 | String key = type2id(alAnot[j].label); |
| 1075 | 83 | boolean isrna = alAnot[j].isValidStruc(); |
| 1076 | ||
| 1077 | 83 | if (isrna) |
| 1078 | { | |
| 1079 | // hardwire to secondary structure if there is RNA secondary | |
| 1080 | // structure on the annotation | |
| 1081 | 66 | key = "SS"; |
| 1082 | } | |
| 1083 | 83 | if (key == null) |
| 1084 | { | |
| 1085 | ||
| 1086 | 4 | continue; |
| 1087 | } | |
| 1088 | ||
| 1089 | // out.append("#=GR "); | |
| 1090 | 79 | out.append(new Format("%-" + maxid + "s").form( |
| 1091 | "#=GR " + printId(seq, jvSuffix) + " " + key + " ")); | |
| 1092 | 79 | ann = alAnot[j].annotations; |
| 1093 | 79 | String sseq = ""; |
| 1094 | 7982 | for (int k = 0, nk = ann.length; k < nk; k++) |
| 1095 | { | |
| 1096 | 7903 | sseq += outputCharacter(key, k, isrna, ann, seq); |
| 1097 | } | |
| 1098 | 79 | out.append(sseq); |
| 1099 | 79 | out.append(newline); |
| 1100 | } | |
| 1101 | } | |
| 1102 | ||
| 1103 | 344 | out.append(new Format("%-" + maxid + "s") |
| 1104 | .form(printId(seq, jvSuffix) + " ")); | |
| 1105 | 344 | out.append(seq.getSequenceAsString()); |
| 1106 | 344 | out.append(newline); |
| 1107 | 344 | i++; |
| 1108 | } | |
| 1109 | ||
| 1110 | // alignment annotation | |
| 1111 | 60 | AlignmentAnnotation aa; |
| 1112 | 60 | AlignmentAnnotation[] an = al.getAlignmentAnnotation(); |
| 1113 | 60 | if (an != null) |
| 1114 | { | |
| 1115 | 272 | for (int ia = 0, na = an.length; ia < na; ia++) |
| 1116 | { | |
| 1117 | 216 | aa = an[ia]; |
| 1118 | 216 | if (aa.autoCalculated || !aa.visible || aa.sequenceRef != null) |
| 1119 | { | |
| 1120 | 211 | continue; |
| 1121 | } | |
| 1122 | 5 | String sseq = ""; |
| 1123 | 5 | String label; |
| 1124 | 5 | String key = ""; |
| 1125 | 5 | if (aa.label.equals("seq")) |
| 1126 | { | |
| 1127 | 1 | label = "seq_cons"; |
| 1128 | } | |
| 1129 | else | |
| 1130 | { | |
| 1131 | 4 | key = type2id(aa.label.toLowerCase(Locale.ROOT)); |
| 1132 | 4 | if (key == null) |
| 1133 | { | |
| 1134 | 0 | label = aa.label; |
| 1135 | } | |
| 1136 | else | |
| 1137 | { | |
| 1138 | 4 | label = key + "_cons"; |
| 1139 | } | |
| 1140 | } | |
| 1141 | 5 | if (label == null) |
| 1142 | { | |
| 1143 | 0 | label = aa.label; |
| 1144 | } | |
| 1145 | 5 | label = label.replace(" ", "_"); |
| 1146 | ||
| 1147 | 5 | out.append( |
| 1148 | new Format("%-" + maxid + "s").form("#=GC " + label + " ")); | |
| 1149 | 5 | boolean isrna = aa.isValidStruc(); |
| 1150 | 453 | for (int j = 0, nj = aa.annotations.length; j < nj; j++) |
| 1151 | { | |
| 1152 | 448 | sseq += outputCharacter(key, j, isrna, aa.annotations, null); |
| 1153 | } | |
| 1154 | 5 | out.append(sseq); |
| 1155 | 5 | out.append(newline); |
| 1156 | } | |
| 1157 | } | |
| 1158 | ||
| 1159 | 60 | out.append("//"); |
| 1160 | 60 | out.append(newline); |
| 1161 | ||
| 1162 | 60 | return out.toString(); |
| 1163 | } | |
| 1164 | ||
| 1165 | /** | |
| 1166 | * add an annotation character to the output row | |
| 1167 | * | |
| 1168 | * @param seq | |
| 1169 | * @param key | |
| 1170 | * @param k | |
| 1171 | * @param isrna | |
| 1172 | * @param ann | |
| 1173 | * @param sequenceI | |
| 1174 | */ | |
| 1175 | 8351 | private char outputCharacter(String key, int k, boolean isrna, |
| 1176 | Annotation[] ann, SequenceI sequenceI) | |
| 1177 | { | |
| 1178 | 8351 | char seq = ' '; |
| 1179 | 8351 | Annotation annot = ann[k]; |
| 1180 | 8351 | String ch = (annot == null) |
| 1181 | 2440 | ? ((sequenceI == null) ? "-" |
| 1182 | : Character.toString(sequenceI.getCharAt(k))) | |
| 1183 | 5911 | : (annot.displayCharacter == null |
| 1184 | ? String.valueOf(annot.secondaryStructure) | |
| 1185 | : annot.displayCharacter); | |
| 1186 | 8351 | if (ch == null) |
| 1187 | { | |
| 1188 | 0 | ch = " "; |
| 1189 | } | |
| 1190 | 8351 | if (key != null && key.equals("SS")) |
| 1191 | { | |
| 1192 | 8134 | char ssannotchar = ' '; |
| 1193 | 8134 | boolean charset = false; |
| 1194 | 8134 | if (annot == null) |
| 1195 | { | |
| 1196 | // sensible gap character | |
| 1197 | 2440 | ssannotchar = ' '; |
| 1198 | 2440 | charset = true; |
| 1199 | } | |
| 1200 | else | |
| 1201 | { | |
| 1202 | // valid secondary structure AND no alternative label (e.g. ' B') | |
| 1203 | 5694 | if (annot.secondaryStructure > ' ' && ch.length() < 2) |
| 1204 | { | |
| 1205 | 3074 | ssannotchar = annot.secondaryStructure; |
| 1206 | 3074 | charset = true; |
| 1207 | } | |
| 1208 | } | |
| 1209 | 8134 | if (charset) |
| 1210 | { | |
| 1211 | 5514 | return (ssannotchar == ' ' && isrna) ? '.' : ssannotchar; |
| 1212 | } | |
| 1213 | } | |
| 1214 | ||
| 1215 | 2837 | if (ch.length() == 0) |
| 1216 | { | |
| 1217 | 0 | seq = '.'; |
| 1218 | } | |
| 1219 | 2837 | else if (ch.length() == 1) |
| 1220 | { | |
| 1221 | 454 | seq = ch.charAt(0); |
| 1222 | } | |
| 1223 | 2383 | else if (ch.length() > 1) |
| 1224 | { | |
| 1225 | 2383 | seq = ch.charAt(1); |
| 1226 | } | |
| 1227 | ||
| 1228 | 2837 | return (seq == ' ' && key != null && key.equals("SS") && isrna) ? '.' |
| 1229 | : seq; | |
| 1230 | } | |
| 1231 | ||
| 1232 | 0 | public String print() |
| 1233 | { | |
| 1234 | 0 | out = new StringBuffer(); |
| 1235 | 0 | out.append("# STOCKHOLM 1.0"); |
| 1236 | 0 | out.append(newline); |
| 1237 | 0 | print(getSeqsAsArray(), false); |
| 1238 | ||
| 1239 | 0 | out.append("//"); |
| 1240 | 0 | out.append(newline); |
| 1241 | 0 | return out.toString(); |
| 1242 | } | |
| 1243 | ||
| 1244 | private static Hashtable typeIds = null; | |
| 1245 | ||
| 1246 | 1 | static |
| 1247 | { | |
| 1248 | 1 | if (typeIds == null) |
| 1249 | { | |
| 1250 | 1 | typeIds = new Hashtable(); |
| 1251 | 1 | typeIds.put("SS", "Secondary Structure"); |
| 1252 | 1 | typeIds.put("SA", "Surface Accessibility"); |
| 1253 | 1 | typeIds.put("TM", "transmembrane"); |
| 1254 | 1 | typeIds.put("PP", "Posterior Probability"); |
| 1255 | 1 | typeIds.put("LI", "ligand binding"); |
| 1256 | 1 | typeIds.put("AS", "active site"); |
| 1257 | 1 | typeIds.put("IN", "intron"); |
| 1258 | 1 | typeIds.put("IR", "interacting residue"); |
| 1259 | 1 | typeIds.put("AC", "accession"); |
| 1260 | 1 | typeIds.put("OS", "organism"); |
| 1261 | 1 | typeIds.put("CL", "class"); |
| 1262 | 1 | typeIds.put("DE", "description"); |
| 1263 | 1 | typeIds.put("DR", "reference"); |
| 1264 | 1 | typeIds.put("LO", "look"); |
| 1265 | 1 | typeIds.put("RF", "Reference Positions"); |
| 1266 | ||
| 1267 | } | |
| 1268 | } | |
| 1269 | ||
| 1270 | 973 | protected static String id2type(String id) |
| 1271 | { | |
| 1272 | 973 | if (typeIds.containsKey(id)) |
| 1273 | { | |
| 1274 | 970 | return (String) typeIds.get(id); |
| 1275 | } | |
| 1276 | 3 | jalview.bin.Console.errPrintln( |
| 1277 | "Warning : Unknown Stockholm annotation type code " + id); | |
| 1278 | 3 | return id; |
| 1279 | } | |
| 1280 | ||
| 1281 | 407 | protected static String type2id(String type) |
| 1282 | { | |
| 1283 | 407 | String key = null; |
| 1284 | 407 | Enumeration e = typeIds.keys(); |
| 1285 | 4494 | while (e.hasMoreElements()) |
| 1286 | { | |
| 1287 | 4490 | Object ll = e.nextElement(); |
| 1288 | 4490 | if (typeIds.get(ll).toString().equalsIgnoreCase(type)) |
| 1289 | { | |
| 1290 | 403 | key = (String) ll; |
| 1291 | 403 | break; |
| 1292 | } | |
| 1293 | } | |
| 1294 | 407 | if (key != null) |
| 1295 | { | |
| 1296 | 403 | return key; |
| 1297 | } | |
| 1298 | 4 | jalview.bin.Console.errPrintln( |
| 1299 | "Warning : Unknown Stockholm annotation type: " + type); | |
| 1300 | 4 | return key; |
| 1301 | } | |
| 1302 | ||
| 1303 | /** | |
| 1304 | * make a friendly ID string. | |
| 1305 | * | |
| 1306 | * @param dataName | |
| 1307 | * @return truncated dataName to after last '/' | |
| 1308 | */ | |
| 1309 | 0 | private String safeName(String dataName) |
| 1310 | { | |
| 1311 | 0 | int b = 0; |
| 1312 | 0 | while ((b = dataName.indexOf("/")) > -1 && b < dataName.length()) |
| 1313 | { | |
| 1314 | 0 | dataName = dataName.substring(b + 1).trim(); |
| 1315 | ||
| 1316 | } | |
| 1317 | 0 | int e = (dataName.length() - dataName.indexOf(".")) + 1; |
| 1318 | 0 | dataName = dataName.substring(1, e).trim(); |
| 1319 | 0 | return dataName; |
| 1320 | } | |
| 1321 | } |