Coverage Report
File ValueAnnotationDistanceModel.java
Coverage histogram
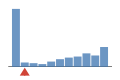
57% of files have more coverage
Classes
| Class | Line # | Actions | |||
|---|---|---|---|---|---|
| ValueAnnotationDistanceModel | 49 | 132 | 58 |
Contributing tests
This file is covered by 3 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | package jalview.analysis.scoremodels; | |
| 22 | ||
| 23 | import jalview.analysis.AlignmentAnnotationUtils; | |
| 24 | import jalview.analysis.AlignmentUtils; | |
| 25 | import jalview.api.AlignmentViewPanel; | |
| 26 | import jalview.api.FeatureRenderer; | |
| 27 | import jalview.api.analysis.ScoreModelI; | |
| 28 | import jalview.api.analysis.SimilarityParamsI; | |
| 29 | import jalview.bin.Console; | |
| 30 | import jalview.datamodel.AlignmentAnnotation; | |
| 31 | import jalview.datamodel.AlignmentView; | |
| 32 | import jalview.datamodel.Annotation; | |
| 33 | import jalview.datamodel.SeqCigar; | |
| 34 | import jalview.datamodel.SequenceI; | |
| 35 | import jalview.math.Matrix; | |
| 36 | import jalview.math.MatrixI; | |
| 37 | import jalview.util.Constants; | |
| 38 | ||
| 39 | import java.util.ArrayList; | |
| 40 | import java.util.HashMap; | |
| 41 | import java.util.HashSet; | |
| 42 | import java.util.List; | |
| 43 | import java.util.Map; | |
| 44 | import java.util.Set; | |
| 45 | ||
| 46 | /* This class contains methods to calculate distance score between | |
| 47 | * secondary structure annotations of the sequences. | |
| 48 | */ | |
| 49 | public class ValueAnnotationDistanceModel extends DistanceScoreModel | |
| 50 | { | |
| 51 | private static final String NAME = "Annotation tracks"; | |
| 52 | ||
| 53 | private String description; | |
| 54 | ||
| 55 | FeatureRenderer fr; | |
| 56 | ||
| 57 | /** | |
| 58 | * Constructor | |
| 59 | */ | |
| 60 | 74 |  public ValueAnnotationDistanceModel() public ValueAnnotationDistanceModel() |
| 61 | { | |
| 62 | ||
| 63 | } | |
| 64 | ||
| 65 | 0 | @Override |
| 66 | public ScoreModelI getInstance(AlignmentViewPanel view) | |
| 67 | { | |
| 68 | 0 | ValueAnnotationDistanceModel instance; |
| 69 | 0 | try |
| 70 | { | |
| 71 | 0 | instance = this.getClass().getDeclaredConstructor().newInstance(); |
| 72 | 0 | instance.configureFromAlignmentView(view); |
| 73 | 0 | return instance; |
| 74 | } catch (InstantiationException | IllegalAccessException e) | |
| 75 | { | |
| 76 | 0 | jalview.bin.Console.errPrintln("Error in " + getClass().getName() |
| 77 | + ".getInstance(): " + e.getMessage()); | |
| 78 | 0 | return null; |
| 79 | } catch (ReflectiveOperationException roe) | |
| 80 | { | |
| 81 | 0 | return null; |
| 82 | } | |
| 83 | } | |
| 84 | ||
| 85 | 0 | boolean configureFromAlignmentView(AlignmentViewPanel view) |
| 86 | ||
| 87 | { | |
| 88 | 0 | fr = view.cloneFeatureRenderer(); |
| 89 | 0 | return true; |
| 90 | } | |
| 91 | ||
| 92 | ArrayList<AlignmentAnnotation> ssForSeqs = null; | |
| 93 | ||
| 94 | 0 | @Override |
| 95 | public SequenceI[] expandSeqData(SequenceI[] sequences, | |
| 96 | AlignmentView seqData, SimilarityParamsI scoreParams, | |
| 97 | List<String> labels, | |
| 98 | ArrayList<AlignmentAnnotation> ssAnnotationForSeqs, | |
| 99 | HashMap<Integer, String> annotationDetails) | |
| 100 | { | |
| 101 | 0 | ssForSeqs = new ArrayList<AlignmentAnnotation>(); |
| 102 | 0 | List<SequenceI> newSequences = new ArrayList<SequenceI>(); |
| 103 | 0 | List<SeqCigar> newCigs = new ArrayList<SeqCigar>(); |
| 104 | 0 | int sq = 0; |
| 105 | ||
| 106 | 0 | AlignmentAnnotation[] alignAnnotList = fr.getViewport().getAlignment() |
| 107 | .getAlignmentAnnotation(); | |
| 108 | ||
| 109 | /* | |
| 110 | * Add secondary structure annotations that are added to the annotation track | |
| 111 | * to the map | |
| 112 | */ | |
| 113 | 0 | Map<SequenceI, ArrayList<AlignmentAnnotation>> ssAlignmentAnnotationForSequences = AlignmentUtils |
| 114 | .getSequenceAssociatedAlignmentAnnotations(alignAnnotList, null, | |
| 115 | true); | |
| 116 | ||
| 117 | 0 | for (SeqCigar scig : seqData.getSequences()) |
| 118 | { | |
| 119 | // get the next sequence that should be bound to this scig: may be null | |
| 120 | 0 | SequenceI alSeq = sequences[sq++]; |
| 121 | 0 | List<AlignmentAnnotation> ssec = ssAlignmentAnnotationForSequences |
| 122 | .get(scig.getRefSeq()); | |
| 123 | ||
| 124 | 0 | if (ssec == null && scoreParams.getExcludeSeqWithoutAnnot()) |
| 125 | { | |
| 126 | 0 | continue; |
| 127 | } | |
| 128 | 0 | else if (ssec == null) |
| 129 | { | |
| 130 | // not defined | |
| 131 | 0 | newSequences.add(alSeq); |
| 132 | 0 | if (alSeq != null) |
| 133 | { | |
| 134 | // labels.add("No Secondary Structure"); | |
| 135 | 0 | labels.add(Constants.STRUCTURE_PROVIDERS.get("None")); |
| 136 | } | |
| 137 | 0 | SeqCigar newSeqCigar = scig; // new SeqCigar(scig); |
| 138 | 0 | newCigs.add(newSeqCigar); |
| 139 | 0 | ssForSeqs.add(null); |
| 140 | } | |
| 141 | else | |
| 142 | { | |
| 143 | 0 | for (int i = 0; i < ssec.size(); i++) |
| 144 | { | |
| 145 | 0 | if (alSeq != null) |
| 146 | { | |
| 147 | // Add annotationDetails if the annotation has | |
| 148 | // ANNOTATION_DETAILS property value (additional metadata) | |
| 149 | ||
| 150 | ||
| 151 | 0 | String provider = ""; |
| 152 | ||
| 153 | 0 | if (ssec.get(i).hasAnnotationDetailsProperty()) |
| 154 | { | |
| 155 | // using key = labels.size() gives the position of the node | |
| 156 | 0 | annotationDetails.put(labels.size(), |
| 157 | ssec.get(i).getAnnotationDetailsProperty()); | |
| 158 | } else { | |
| 159 | 0 | annotationDetails.put(labels.size(), |
| 160 | 0 | ssec.get(i).label+(ssec.get(i).description!=null ? ssec.get(i).description:"")); |
| 161 | 0 | provider = alSeq.getName(); |
| 162 | } | |
| 163 | // .extractSSSourceFromAnnotationDescription(ssec.get(i)); | |
| 164 | 0 | labels.add(provider); |
| 165 | } | |
| 166 | 0 | newSequences.add(alSeq); |
| 167 | 0 | SeqCigar newSeqCigar = scig; // new SeqCigar(scig); |
| 168 | 0 | newCigs.add(newSeqCigar); |
| 169 | 0 | ssForSeqs.add(ssec.get(i)); |
| 170 | } | |
| 171 | } | |
| 172 | } | |
| 173 | 0 | ssAnnotationForSeqs.addAll(ssForSeqs); |
| 174 | 0 | seqData.setSequences(newCigs.toArray(new SeqCigar[0])); |
| 175 | 0 | return newSequences.toArray(new SequenceI[0]); |
| 176 | ||
| 177 | } | |
| 178 | ||
| 179 | ||
| 180 | boolean hamming=false; // one not equals other | |
| 181 | boolean logIt = false; // log1p scale before difference calc | |
| 182 | boolean relDiff = true; // compute difference of normalised values w.r.t. each rows max/min | |
| 183 | ||
| 184 | 0 | protected void configureMode(boolean hamming, boolean logIt, boolean relDiff) { |
| 185 | 0 | this.hamming=hamming; |
| 186 | 0 | this.logIt=logIt; |
| 187 | 0 | this.relDiff=relDiff; |
| 188 | } | |
| 189 | ||
| 190 | /** | |
| 191 | * Calculates distance score [i][j] between each pair of protein sequences | |
| 192 | * based on their secondary structure annotations (H, E, C). The final score | |
| 193 | * is normalised by the number of alignment columns processed, providing an | |
| 194 | * average similarity score. | |
| 195 | * <p> | |
| 196 | * The parameters argument can include settings for handling gap-residue | |
| 197 | * aligned positions and may determine if the score calculation is based on | |
| 198 | * the longer or shorter sequence in each pair. This can be important for | |
| 199 | * handling partial alignments or sequences of significantly different | |
| 200 | * lengths. | |
| 201 | * | |
| 202 | * @param seqData | |
| 203 | * The aligned sequence data including secondary structure | |
| 204 | * annotations. | |
| 205 | * @param params | |
| 206 | * Additional parameters for customising the scoring process, such as | |
| 207 | * gap handling and sequence length consideration. | |
| 208 | */ | |
| 209 | 0 | @Override |
| 210 | public MatrixI findDistances(AlignmentView seqData, | |
| 211 | SimilarityParamsI params) | |
| 212 | { | |
| 213 | 0 | configureMode(false,true,true); |
| 214 | 0 | Console.trace("Starting to calculate: "+getDescription()); |
| 215 | 0 | if (ssForSeqs == null |
| 216 | || ssForSeqs.size() != seqData.getSequences().length) | |
| 217 | { | |
| 218 | // expandSeqData needs to be called to initialise the hash | |
| 219 | 0 | SequenceI[] sequences = new SequenceI[seqData.getSequences().length]; |
| 220 | // we throw away the new labels in this case.. | |
| 221 | 0 | expandSeqData(sequences, seqData, params, new ArrayList<String>(), |
| 222 | new ArrayList<AlignmentAnnotation>(), | |
| 223 | new HashMap<Integer, String>()); | |
| 224 | } | |
| 225 | 0 | SeqCigar[] seqs = seqData.getSequences(); |
| 226 | 0 | int noseqs = seqs.length; // no of sequences |
| 227 | 0 | int cpwidth = 0; |
| 228 | 0 | double[][] differences = new double[noseqs][noseqs]; // matrix to store |
| 229 | // similarity score | |
| 230 | // secondary structure source parameter selected by the user from the drop | |
| 231 | // down. | |
| 232 | 0 | String ssSource = params.getSecondaryStructureSource(); |
| 233 | 0 | if (ssSource == null || ssSource == "") |
| 234 | { | |
| 235 | 0 | ssSource = Constants.SS_ALL_PROVIDERS; |
| 236 | } | |
| 237 | ||
| 238 | // need to get real position for view position | |
| 239 | 0 | int[] viscont = seqData.getVisibleContigs(); |
| 240 | ||
| 241 | /* | |
| 242 | * scan each column, compute and add to each similarity[i, j] | |
| 243 | * the number of secondary structure annotation that seqi | |
| 244 | * and seqj do not share | |
| 245 | */ | |
| 246 | 0 | double maxDiff = 0; |
| 247 | 0 | float maxVal=0f; |
| 248 | 0 | for (int i = 0; i < noseqs; i++) |
| 249 | { | |
| 250 | 0 | AlignmentAnnotation aa_i = ssForSeqs.get(i); |
| 251 | //if (aa_i.graphMax>22000) { continue; } | |
| 252 | 0 | maxVal = Math.max(maxVal, aa_i.graphMax); |
| 253 | 0 | maxDiff = Math.max(maxDiff, |
| 254 | Math.abs(aa_i.graphMax - Math.min(0, aa_i.graphMin))); | |
| 255 | } | |
| 256 | // for (int i=0;i < noseqs;i++) | |
| 257 | // { | |
| 258 | // AlignmentAnnotation aa_i = ssForSeqs.get(i); | |
| 259 | // aa_i.graphMin=0f; | |
| 260 | // aa_i.graphMax=maxVal; | |
| 261 | // } | |
| 262 | 0 | if (logIt) |
| 263 | { | |
| 264 | 0 | maxDiff = Math.log1p(maxDiff); |
| 265 | } | |
| 266 | 0 | Annotation ann1; |
| 267 | 0 | Annotation ann2; |
| 268 | ||
| 269 | 0 | for (int vc = 0; vc < viscont.length; vc += 2) |
| 270 | { | |
| 271 | // Iterates for each column position | |
| 272 | 0 | for (int cpos = viscont[vc]; cpos <= viscont[vc + 1]; cpos++) |
| 273 | { | |
| 274 | 0 | cpwidth++; // used to normalise the similarity score |
| 275 | ||
| 276 | /* | |
| 277 | * get set of sequences without gap in the current column | |
| 278 | */ | |
| 279 | 0 | Set<SeqCigar> seqsWithoutGapAtCol = findSeqsWithoutGapAtColumn(seqs, |
| 280 | cpos); | |
| 281 | ||
| 282 | /* | |
| 283 | * calculate similarity score for each secondary structure annotation on i'th and j'th | |
| 284 | * sequence and add this measure to the similarities matrix | |
| 285 | * for [i, j] for j > i | |
| 286 | */ | |
| 287 | ||
| 288 | 0 | for (int i = 0; i < (noseqs - 1); i++) |
| 289 | { | |
| 290 | 0 | AlignmentAnnotation aa_i = ssForSeqs.get(i); |
| 291 | 0 | boolean undefinedSS1 = aa_i == null; |
| 292 | // check if the sequence contains gap in the current column | |
| 293 | 0 | boolean gap1 = !seqsWithoutGapAtCol.contains(seqs[i]); |
| 294 | // secondary structure is fetched only if the current column is not | |
| 295 | // gap for the sequence | |
| 296 | 0 | double ss1 = 0; |
| 297 | 0 | if (!gap1 && !undefinedSS1) |
| 298 | { | |
| 299 | // fetch the position in sequence for the column and finds the | |
| 300 | // corresponding secondary structure annotation | |
| 301 | // TO DO - consider based on priority and displayed | |
| 302 | 0 | int seqPosition_i = seqs[i].findPosition(cpos); |
| 303 | 0 | if (aa_i != null) |
| 304 | { | |
| 305 | 0 | ann1 = aa_i.getAnnotationForPosition(seqPosition_i); |
| 306 | 0 | if (ann1 != null) |
| 307 | { | |
| 308 | 0 | ss1 = ann1.value; |
| 309 | 0 | if (relDiff) { |
| 310 | 0 | ss1 = ss1/aa_i.graphMax; |
| 311 | } | |
| 312 | 0 | if (logIt) |
| 313 | { | |
| 314 | 0 | ss1 = Math.log1p(ss1); |
| 315 | } | |
| 316 | } | |
| 317 | } | |
| 318 | } | |
| 319 | // Iterates for each sequences | |
| 320 | 0 | for (int j = i + 1; j < noseqs; j++) |
| 321 | { | |
| 322 | ||
| 323 | // check if ss is defined | |
| 324 | 0 | AlignmentAnnotation aa_j = ssForSeqs.get(j); |
| 325 | ||
| 326 | 0 | boolean undefinedSS2 = aa_j == null; |
| 327 | ||
| 328 | // Set similarity to max score if both SS are not defined | |
| 329 | 0 | if (undefinedSS1 || undefinedSS2) |
| 330 | { | |
| 331 | 0 | differences[i][j] += 0; |
| 332 | 0 | continue; |
| 333 | } | |
| 334 | ||
| 335 | // Set similarity to minimum score if either one SS is not defined | |
| 336 | // else if (undefinedSS1 || undefinedSS2) | |
| 337 | // { | |
| 338 | // differences[i][j] += maxDiff; | |
| 339 | // continue; | |
| 340 | // } | |
| 341 | ||
| 342 | 0 | boolean gap2 = !seqsWithoutGapAtCol.contains(seqs[j]); |
| 343 | ||
| 344 | // Variable to store secondary structure at the current column | |
| 345 | 0 | double ss2 = 0; |
| 346 | ||
| 347 | 0 | if (!gap2 && !undefinedSS2) |
| 348 | { | |
| 349 | 0 | int seqPosition = seqs[j].findPosition(cpos); |
| 350 | 0 | ann2 = aa_j.getAnnotationForPosition(seqPosition); |
| 351 | 0 | if (ann2 != null) |
| 352 | { | |
| 353 | 0 | ss2 = ann2.value; |
| 354 | 0 | if (relDiff) { |
| 355 | 0 | ss2 = ss2/aa_j.graphMax; |
| 356 | } | |
| 357 | 0 | if (logIt) |
| 358 | { | |
| 359 | 0 | ss2 = Math.log1p(ss2); |
| 360 | } | |
| 361 | } | |
| 362 | } | |
| 363 | ||
| 364 | 0 | if ((!gap1 && !gap2)) |
| 365 | { | |
| 366 | 0 | if (hamming) |
| 367 | { | |
| 368 | 0 | differences[i][j]+=(ss1==ss2 ? 0:1); |
| 369 | } | |
| 370 | else | |
| 371 | { | |
| 372 | // Calculate similarity score based on the substitution matrix | |
| 373 | 0 | double similarityScore = Math.abs(ss1 - ss2); |
| 374 | 0 | if (logIt) |
| 375 | { | |
| 376 | 0 | similarityScore = Math.log1p(similarityScore); |
| 377 | } | |
| 378 | 0 | differences[i][j] += similarityScore; |
| 379 | } | |
| 380 | } | |
| 381 | else | |
| 382 | { | |
| 383 | 0 | if (hamming) {differences[i][j]+=1;} |
| 384 | else { | |
| 385 | 0 | differences[i][j] += 0;// maxDiff; } |
| 386 | } | |
| 387 | } | |
| 388 | } | |
| 389 | } | |
| 390 | } | |
| 391 | } | |
| 392 | ||
| 393 | /* | |
| 394 | * normalise the similarity scores (summed over columns) by the | |
| 395 | * number of visible columns used in the calculation | |
| 396 | * and fill in the bottom half of the matrix | |
| 397 | */ | |
| 398 | // TODO JAL-2424 cpwidth may be out by 1 - affects scores but not tree shape | |
| 399 | ||
| 400 | 0 | for (int i = 0; i < noseqs; i++) |
| 401 | { | |
| 402 | 0 | for (int j = i + 1; j < noseqs; j++) |
| 403 | { | |
| 404 | 0 | differences[i][j] /= cpwidth; |
| 405 | 0 | differences[j][i] = differences[i][j]; |
| 406 | } | |
| 407 | } | |
| 408 | 0 | return (new Matrix(differences)); |
| 409 | ||
| 410 | } | |
| 411 | ||
| 412 | /** | |
| 413 | * Builds and returns a set containing sequences (SeqCigar) which do not have | |
| 414 | * a gap at the given column position. | |
| 415 | * | |
| 416 | * @param seqs | |
| 417 | * @param columnPosition | |
| 418 | * (0..) | |
| 419 | * @return | |
| 420 | */ | |
| 421 | 0 | private Set<SeqCigar> findSeqsWithoutGapAtColumn(SeqCigar[] seqs, |
| 422 | int columnPosition) | |
| 423 | { | |
| 424 | 0 | Set<SeqCigar> seqsWithoutGapAtCol = new HashSet<>(); |
| 425 | 0 | for (SeqCigar seq : seqs) |
| 426 | { | |
| 427 | 0 | int spos = seq.findPosition(columnPosition); |
| 428 | 0 | if (spos != -1) |
| 429 | { | |
| 430 | /* | |
| 431 | * position is not a gap | |
| 432 | */ | |
| 433 | 0 | seqsWithoutGapAtCol.add(seq); |
| 434 | } | |
| 435 | } | |
| 436 | 0 | return seqsWithoutGapAtCol; |
| 437 | } | |
| 438 | ||
| 439 | 148 | @Override |
| 440 | public String getName() | |
| 441 | { | |
| 442 | 148 | return "Annotation"; |
| 443 | } | |
| 444 | ||
| 445 | 0 | @Override |
| 446 | public String getDescription() | |
| 447 | { | |
| 448 | 0 | return "Score between annotation ("+(hamming?"hamming":(logIt?"log1p ":"")+(relDiff?"Relative":",Absolute"))+")"; |
| 449 | } | |
| 450 | ||
| 451 | 4 | @Override |
| 452 | public boolean isDNA() | |
| 453 | { | |
| 454 | 4 | return false; |
| 455 | } | |
| 456 | ||
| 457 | 2 | @Override |
| 458 | public boolean isProtein() | |
| 459 | { | |
| 460 | 2 | return false; |
| 461 | } | |
| 462 | ||
| 463 | 6 | @Override |
| 464 | public boolean isSecondaryStructure() | |
| 465 | { | |
| 466 | 6 | return true; |
| 467 | } | |
| 468 | ||
| 469 | 0 | @Override |
| 470 | public String toString() | |
| 471 | { | |
| 472 | 0 | return "Score between sequences based on similarity between binary " |
| 473 | + "vectors marking secondary structure displayed at each column"; | |
| 474 | } | |
| 475 | } |