Coverage Report
File PIRFile.java
Coverage histogram
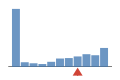
29% of files have more coverage
Classes
| Class | Line # | Actions | |||
|---|---|---|---|---|---|
| PIRFile | 30 | 69 | 24 |
Contributing tests
This file is covered by 1 test.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | package jalview.io; | |
| 22 | ||
| 23 | import jalview.datamodel.Sequence; | |
| 24 | import jalview.datamodel.SequenceI; | |
| 25 | import jalview.util.Comparison; | |
| 26 | ||
| 27 | import java.io.IOException; | |
| 28 | import java.util.Vector; | |
| 29 | ||
| 30 | public class PIRFile extends AlignFile | |
| 31 | { | |
| 32 | public static boolean useModellerOutput = false; | |
| 33 | ||
| 34 | Vector words = new Vector(); // Stores the words in a line after splitting | |
| 35 | ||
| 36 | 3 |  public PIRFile() public PIRFile() |
| 37 | { | |
| 38 | } | |
| 39 | ||
| 40 | 0 | public PIRFile(String inFile, DataSourceType sourceType) |
| 41 | throws IOException | |
| 42 | { | |
| 43 | 0 | super(inFile, sourceType); |
| 44 | } | |
| 45 | ||
| 46 | 1 | public PIRFile(FileParse source) throws IOException |
| 47 | { | |
| 48 | 1 | super(source); |
| 49 | } | |
| 50 | ||
| 51 | 1 | @Override |
| 52 | public void parse() throws IOException | |
| 53 | { | |
| 54 | 1 | StringBuffer sequence; |
| 55 | 1 | String line = null; |
| 56 | 1 | ModellerDescription md; |
| 57 | ||
| 58 | ? | while ((line = nextLine()) != null) |
| 59 | { | |
| 60 | 15 | if (line.length() == 0) |
| 61 | { | |
| 62 | // jalview.bin.Console.outPrintln("blank line"); | |
| 63 | 0 | continue; |
| 64 | } | |
| 65 | 15 | if (line.indexOf("C;") == 0 || line.indexOf("#") == 0) |
| 66 | { | |
| 67 | 0 | continue; |
| 68 | } | |
| 69 | 15 | Sequence newSeq = parseId(line.substring(line.indexOf(";") + 1)); |
| 70 | ||
| 71 | 15 | sequence = new StringBuffer(); |
| 72 | ||
| 73 | 15 | newSeq.setDescription(nextLine()); // this is the title line |
| 74 | ||
| 75 | 15 | boolean starFound = false; |
| 76 | ||
| 77 | 60 | while (!starFound) |
| 78 | { | |
| 79 | 45 | line = nextLine(); |
| 80 | 45 | sequence.append(line); |
| 81 | ||
| 82 | 45 | if (line == null) |
| 83 | { | |
| 84 | 0 | break; |
| 85 | } | |
| 86 | ||
| 87 | 45 | if (line.indexOf("*") > -1) |
| 88 | { | |
| 89 | 15 | starFound = true; |
| 90 | } | |
| 91 | } | |
| 92 | ||
| 93 | 15 | if (sequence.length() > 0) |
| 94 | { | |
| 95 | 15 | sequence.setLength(sequence.length() - 1); |
| 96 | 15 | newSeq.setSequence(sequence.toString()); |
| 97 | ||
| 98 | 15 | seqs.addElement(newSeq); |
| 99 | ||
| 100 | 15 | md = new ModellerDescription(newSeq.getDescription()); |
| 101 | 15 | md.updateSequenceI(newSeq); |
| 102 | } | |
| 103 | } | |
| 104 | } | |
| 105 | ||
| 106 | 3 | @Override |
| 107 | public String print(SequenceI[] s, boolean jvsuffix) | |
| 108 | { | |
| 109 | 3 | boolean is_NA = Comparison.isNucleotide(s); |
| 110 | 3 | int len = 72; |
| 111 | 3 | StringBuffer out = new StringBuffer(); |
| 112 | 3 | int i = 0; |
| 113 | 3 | ModellerDescription md; |
| 114 | ||
| 115 | 48 | while ((i < s.length) && (s[i] != null)) |
| 116 | { | |
| 117 | 45 | String seq = s[i].getSequenceAsString(); |
| 118 | 45 | seq = seq + "*"; |
| 119 | ||
| 120 | 45 | if (is_NA) |
| 121 | { | |
| 122 | // modeller doesn't really do nucleotides, so we don't do anything fancy | |
| 123 | // Official tags area as follows, for now we'll use P1 and DL | |
| 124 | // Protein (complete) P1 | |
| 125 | // Protein (fragment) F1 | |
| 126 | // DNA (linear) Dl | |
| 127 | // DNA (circular) DC | |
| 128 | // RNA (linear) RL | |
| 129 | // RNA (circular) RC | |
| 130 | // tRNA N3 | |
| 131 | // other functional RNA N1 | |
| 132 | ||
| 133 | 0 | out.append(">N1;" + s[i].getName()); |
| 134 | 0 | out.append(newline); |
| 135 | 0 | if (s[i].getDescription() == null) |
| 136 | { | |
| 137 | 0 | out.append(s[i].getName() + " " |
| 138 | + (s[i].getEnd() - s[i].getStart() + 1)); | |
| 139 | 0 | out.append(is_NA ? " bases" : " residues"); |
| 140 | 0 | out.append(newline); |
| 141 | } | |
| 142 | else | |
| 143 | { | |
| 144 | 0 | out.append(s[i].getDescription()); |
| 145 | 0 | out.append(newline); |
| 146 | } | |
| 147 | } | |
| 148 | else | |
| 149 | { | |
| 150 | ||
| 151 | 45 | if (useModellerOutput) |
| 152 | { | |
| 153 | 0 | out.append(">P1;" + s[i].getName()); |
| 154 | 0 | out.append(newline); |
| 155 | 0 | md = new ModellerDescription(s[i]); |
| 156 | 0 | out.append(md.getDescriptionLine()); |
| 157 | 0 | out.append(newline); |
| 158 | } | |
| 159 | else | |
| 160 | { | |
| 161 | 45 | out.append(">P1;" + printId(s[i], jvsuffix)); |
| 162 | 45 | out.append(newline); |
| 163 | 45 | if (s[i].getDescription() != null) |
| 164 | { | |
| 165 | 45 | out.append(s[i].getDescription()); |
| 166 | 45 | out.append(newline); |
| 167 | } | |
| 168 | else | |
| 169 | { | |
| 170 | 0 | out.append(s[i].getName() + " " |
| 171 | + (s[i].getEnd() - s[i].getStart() + 1) + " residues"); | |
| 172 | 0 | out.append(newline); |
| 173 | } | |
| 174 | } | |
| 175 | } | |
| 176 | 45 | int nochunks = (seq.length() / len) |
| 177 | 45 | + (seq.length() % len > 0 ? 1 : 0); |
| 178 | ||
| 179 | 180 | for (int j = 0; j < nochunks; j++) |
| 180 | { | |
| 181 | 135 | int start = j * len; |
| 182 | 135 | int end = start + len; |
| 183 | ||
| 184 | 135 | if (end < seq.length()) |
| 185 | { | |
| 186 | 90 | out.append(seq.substring(start, end)); |
| 187 | 90 | out.append(newline); |
| 188 | } | |
| 189 | 45 | else if (start < seq.length()) |
| 190 | { | |
| 191 | 45 | out.append(seq.substring(start)); |
| 192 | 45 | out.append(newline); |
| 193 | } | |
| 194 | } | |
| 195 | ||
| 196 | 45 | i++; |
| 197 | } | |
| 198 | ||
| 199 | 3 | return out.toString(); |
| 200 | } | |
| 201 | ||
| 202 | } |