Coverage Report
File AlignmentUtils.java
Coverage histogram
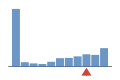
21% of files have more coverage
Classes
| Class | Line # | Actions | |||
|---|---|---|---|---|---|
| AlignmentUtils | 86 | 928 | 352 | ||
| AlignmentUtils.DnaVariant | 102 | 6 | 6 |
Contributing tests
This file is covered by 284 tests.
.
Source view
| 1 | /* | |
| 2 | * Jalview - A Sequence Alignment Editor and Viewer ($$Version-Rel$$) | |
| 3 | * Copyright (C) $$Year-Rel$$ The Jalview Authors | |
| 4 | * | |
| 5 | * This file is part of Jalview. | |
| 6 | * | |
| 7 | * Jalview is free software: you can redistribute it and/or | |
| 8 | * modify it under the terms of the GNU General Public License | |
| 9 | * as published by the Free Software Foundation, either version 3 | |
| 10 | * of the License, or (at your option) any later version. | |
| 11 | * | |
| 12 | * Jalview is distributed in the hope that it will be useful, but | |
| 13 | * WITHOUT ANY WARRANTY; without even the implied warranty | |
| 14 | * of MERCHANTABILITY or FITNESS FOR A PARTICULAR | |
| 15 | * PURPOSE. See the GNU General Public License for more details. | |
| 16 | * | |
| 17 | * You should have received a copy of the GNU General Public License | |
| 18 | * along with Jalview. If not, see <http://www.gnu.org/licenses/>. | |
| 19 | * The Jalview Authors are detailed in the 'AUTHORS' file. | |
| 20 | */ | |
| 21 | package jalview.analysis; | |
| 22 | ||
| 23 | import java.awt.Color; | |
| 24 | import java.util.ArrayList; | |
| 25 | import java.util.Arrays; | |
| 26 | import java.util.Collection; | |
| 27 | import java.util.Collections; | |
| 28 | import java.util.HashMap; | |
| 29 | import java.util.HashSet; | |
| 30 | import java.util.Iterator; | |
| 31 | import java.util.LinkedHashMap; | |
| 32 | import java.util.List; | |
| 33 | import java.util.Locale; | |
| 34 | import java.util.Map; | |
| 35 | import java.util.Map.Entry; | |
| 36 | import java.util.NoSuchElementException; | |
| 37 | import java.util.Set; | |
| 38 | import java.util.SortedMap; | |
| 39 | import java.util.TreeMap; | |
| 40 | import java.util.Vector; | |
| 41 | import java.util.stream.Collectors; | |
| 42 | ||
| 43 | import org.jcolorbrewer.ColorBrewer; | |
| 44 | ||
| 45 | import jalview.api.AlignCalcWorkerI; | |
| 46 | import jalview.bin.Console; | |
| 47 | import jalview.commands.RemoveGapColCommand; | |
| 48 | import jalview.datamodel.AlignedCodon; | |
| 49 | import jalview.datamodel.AlignedCodonFrame; | |
| 50 | import jalview.datamodel.AlignedCodonFrame.SequenceToSequenceMapping; | |
| 51 | import jalview.datamodel.Alignment; | |
| 52 | import jalview.datamodel.AlignmentAnnotation; | |
| 53 | import jalview.datamodel.AlignmentI; | |
| 54 | import jalview.datamodel.Annotation; | |
| 55 | import jalview.datamodel.ContactMatrixI; | |
| 56 | import jalview.datamodel.DBRefEntry; | |
| 57 | import jalview.datamodel.GeneLociI; | |
| 58 | import jalview.datamodel.IncompleteCodonException; | |
| 59 | import jalview.datamodel.Mapping; | |
| 60 | import jalview.datamodel.PDBEntry; | |
| 61 | import jalview.datamodel.SeqCigar; | |
| 62 | import jalview.datamodel.Sequence; | |
| 63 | import jalview.datamodel.SequenceFeature; | |
| 64 | import jalview.datamodel.SequenceGroup; | |
| 65 | import jalview.datamodel.SequenceI; | |
| 66 | import jalview.datamodel.features.SequenceFeatures; | |
| 67 | import jalview.gui.AlignmentPanel; | |
| 68 | import jalview.io.gff.SequenceOntologyI; | |
| 69 | import jalview.schemes.ResidueProperties; | |
| 70 | import jalview.util.Comparison; | |
| 71 | import jalview.util.Constants; | |
| 72 | import jalview.util.DBRefUtils; | |
| 73 | import jalview.util.IntRangeComparator; | |
| 74 | import jalview.util.MapList; | |
| 75 | import jalview.util.MappingUtils; | |
| 76 | import jalview.util.MessageManager; | |
| 77 | import jalview.workers.SecondaryStructureConsensusThread; | |
| 78 | ||
| 79 | /** | |
| 80 | * grab bag of useful alignment manipulation operations Expect these to be | |
| 81 | * refactored elsewhere at some point. | |
| 82 | * | |
| 83 | * @author jimp | |
| 84 | * | |
| 85 | */ | |
| 86 | public class AlignmentUtils | |
| 87 | { | |
| 88 | private static final int CODON_LENGTH = 3; | |
| 89 | ||
| 90 | private static final String SEQUENCE_VARIANT = "sequence_variant:"; | |
| 91 | ||
| 92 | /* | |
| 93 | * the 'id' attribute is provided for variant features fetched from | |
| 94 | * Ensembl using its REST service with JSON format | |
| 95 | */ | |
| 96 | public static final String VARIANT_ID = "id"; | |
| 97 | ||
| 98 | /** | |
| 99 | * A data model to hold the 'normal' base value at a position, and an optional | |
| 100 | * sequence variant feature | |
| 101 | */ | |
| 102 | static final class DnaVariant | |
| 103 | { | |
| 104 | final String base; | |
| 105 | ||
| 106 | SequenceFeature variant; | |
| 107 | ||
| 108 | 0 |  DnaVariant(String nuc) DnaVariant(String nuc) |
| 109 | { | |
| 110 | 0 | base = nuc; |
| 111 | 0 | variant = null; |
| 112 | } | |
| 113 | ||
| 114 | 0 | DnaVariant(String nuc, SequenceFeature var) |
| 115 | { | |
| 116 | 0 | base = nuc; |
| 117 | 0 | variant = var; |
| 118 | } | |
| 119 | ||
| 120 | 0 | public String getSource() |
| 121 | { | |
| 122 | 0 | return variant == null ? null : variant.getFeatureGroup(); |
| 123 | } | |
| 124 | ||
| 125 | /** | |
| 126 | * toString for aid in the debugger only | |
| 127 | */ | |
| 128 | 0 | @Override |
| 129 | public String toString() | |
| 130 | { | |
| 131 | 0 | return base + ":" + (variant == null ? "" : variant.getDescription()); |
| 132 | } | |
| 133 | } | |
| 134 | ||
| 135 | /** | |
| 136 | * given an existing alignment, create a new alignment including all, or up to | |
| 137 | * flankSize additional symbols from each sequence's dataset sequence | |
| 138 | * | |
| 139 | * @param core | |
| 140 | * @param flankSize | |
| 141 | * @return AlignmentI | |
| 142 | */ | |
| 143 | 27 | public static AlignmentI expandContext(AlignmentI core, int flankSize) |
| 144 | { | |
| 145 | 27 | List<SequenceI> sq = new ArrayList<>(); |
| 146 | 27 | int maxoffset = 0; |
| 147 | 27 | for (SequenceI s : core.getSequences()) |
| 148 | { | |
| 149 | 131 | SequenceI newSeq = s.deriveSequence(); |
| 150 | 131 | final int newSeqStart = newSeq.getStart() - 1; |
| 151 | 131 | if (newSeqStart > maxoffset |
| 152 | && newSeq.getDatasetSequence().getStart() < s.getStart()) | |
| 153 | { | |
| 154 | 131 | maxoffset = newSeqStart; |
| 155 | } | |
| 156 | 131 | sq.add(newSeq); |
| 157 | } | |
| 158 | 27 | if (flankSize > -1) |
| 159 | { | |
| 160 | 25 | maxoffset = Math.min(maxoffset, flankSize); |
| 161 | } | |
| 162 | ||
| 163 | /* | |
| 164 | * now add offset left and right to create an expanded alignment | |
| 165 | */ | |
| 166 | 27 | for (SequenceI s : sq) |
| 167 | { | |
| 168 | 131 | SequenceI ds = s; |
| 169 | 262 | while (ds.getDatasetSequence() != null) |
| 170 | { | |
| 171 | 131 | ds = ds.getDatasetSequence(); |
| 172 | } | |
| 173 | 131 | int s_end = s.findPosition(s.getStart() + s.getLength()); |
| 174 | // find available flanking residues for sequence | |
| 175 | 131 | int ustream_ds = s.getStart() - ds.getStart(); |
| 176 | 131 | int dstream_ds = ds.getEnd() - s_end; |
| 177 | ||
| 178 | // build new flanked sequence | |
| 179 | ||
| 180 | // compute gap padding to start of flanking sequence | |
| 181 | 131 | int offset = maxoffset - ustream_ds; |
| 182 | ||
| 183 | // padding is gapChar x ( maxoffset - min(ustream_ds, flank) | |
| 184 | 131 | if (flankSize >= 0) |
| 185 | { | |
| 186 | 125 | if (flankSize < ustream_ds) |
| 187 | { | |
| 188 | // take up to flankSize residues | |
| 189 | 40 | offset = maxoffset - flankSize; |
| 190 | 40 | ustream_ds = flankSize; |
| 191 | } | |
| 192 | 125 | if (flankSize <= dstream_ds) |
| 193 | { | |
| 194 | 116 | dstream_ds = flankSize - 1; |
| 195 | } | |
| 196 | } | |
| 197 | // TODO use Character.toLowerCase to avoid creating String objects? | |
| 198 | 131 | char[] upstream = new String(ds |
| 199 | .getSequence(s.getStart() - 1 - ustream_ds, s.getStart() - 1)) | |
| 200 | .toLowerCase(Locale.ROOT).toCharArray(); | |
| 201 | 131 | char[] downstream = new String( |
| 202 | ds.getSequence(s_end - 1, s_end + dstream_ds)) | |
| 203 | .toLowerCase(Locale.ROOT).toCharArray(); | |
| 204 | 131 | char[] coreseq = s.getSequence(); |
| 205 | 131 | char[] nseq = new char[offset + upstream.length + downstream.length |
| 206 | + coreseq.length]; | |
| 207 | 131 | char c = core.getGapCharacter(); |
| 208 | ||
| 209 | 131 | int p = 0; |
| 210 | 461 | for (; p < offset; p++) |
| 211 | { | |
| 212 | 330 | nseq[p] = c; |
| 213 | } | |
| 214 | ||
| 215 | 131 | System.arraycopy(upstream, 0, nseq, p, upstream.length); |
| 216 | 131 | System.arraycopy(coreseq, 0, nseq, p + upstream.length, |
| 217 | coreseq.length); | |
| 218 | 131 | System.arraycopy(downstream, 0, nseq, |
| 219 | p + coreseq.length + upstream.length, downstream.length); | |
| 220 | 131 | s.setSequence(new String(nseq)); |
| 221 | 131 | s.setStart(s.getStart() - ustream_ds); |
| 222 | 131 | s.setEnd(s_end + downstream.length); |
| 223 | } | |
| 224 | 27 | AlignmentI newAl = new jalview.datamodel.Alignment( |
| 225 | sq.toArray(new SequenceI[0])); | |
| 226 | 27 | for (SequenceI s : sq) |
| 227 | { | |
| 228 | 131 | if (s.getAnnotation() != null) |
| 229 | { | |
| 230 | 1 | for (AlignmentAnnotation aa : s.getAnnotation()) |
| 231 | { | |
| 232 | 1 | aa.adjustForAlignment(); // JAL-1712 fix |
| 233 | 1 | newAl.addAnnotation(aa); |
| 234 | } | |
| 235 | } | |
| 236 | } | |
| 237 | 27 | newAl.setDataset(core.getDataset()); |
| 238 | 27 | return newAl; |
| 239 | } | |
| 240 | ||
| 241 | /** | |
| 242 | * Returns the index (zero-based position) of a sequence in an alignment, or | |
| 243 | * -1 if not found. | |
| 244 | * | |
| 245 | * @param al | |
| 246 | * @param seq | |
| 247 | * @return | |
| 248 | */ | |
| 249 | 59568 | public static int getSequenceIndex(AlignmentI al, SequenceI seq) |
| 250 | { | |
| 251 | 59568 | int result = -1; |
| 252 | 59568 | int pos = 0; |
| 253 | 59568 | for (SequenceI alSeq : al.getSequences()) |
| 254 | { | |
| 255 | 126272048 | if (alSeq == seq) |
| 256 | { | |
| 257 | 59510 | result = pos; |
| 258 | 59510 | break; |
| 259 | } | |
| 260 | 126212538 | pos++; |
| 261 | } | |
| 262 | 59568 | return result; |
| 263 | } | |
| 264 | ||
| 265 | /** | |
| 266 | * Returns a map of lists of sequences in the alignment, keyed by sequence | |
| 267 | * name. For use in mapping between different alignment views of the same | |
| 268 | * sequences. | |
| 269 | * | |
| 270 | * @see jalview.datamodel.AlignmentI#getSequencesByName() | |
| 271 | */ | |
| 272 | 1 | public static Map<String, List<SequenceI>> getSequencesByName( |
| 273 | AlignmentI al) | |
| 274 | { | |
| 275 | 1 | Map<String, List<SequenceI>> theMap = new LinkedHashMap<>(); |
| 276 | 1 | for (SequenceI seq : al.getSequences()) |
| 277 | { | |
| 278 | 3 | String name = seq.getName(); |
| 279 | 3 | if (name != null) |
| 280 | { | |
| 281 | 3 | List<SequenceI> seqs = theMap.get(name); |
| 282 | 3 | if (seqs == null) |
| 283 | { | |
| 284 | 2 | seqs = new ArrayList<>(); |
| 285 | 2 | theMap.put(name, seqs); |
| 286 | } | |
| 287 | 3 | seqs.add(seq); |
| 288 | } | |
| 289 | } | |
| 290 | 1 | return theMap; |
| 291 | } | |
| 292 | ||
| 293 | /** | |
| 294 | * Build mapping of protein to cDNA alignment. Mappings are made between | |
| 295 | * sequences where the cDNA translates to the protein sequence. Any new | |
| 296 | * mappings are added to the protein alignment. Returns true if any mappings | |
| 297 | * either already exist or were added, else false. | |
| 298 | * | |
| 299 | * @param proteinAlignment | |
| 300 | * @param cdnaAlignment | |
| 301 | * @return | |
| 302 | */ | |
| 303 | 4 | public static boolean mapProteinAlignmentToCdna( |
| 304 | final AlignmentI proteinAlignment, final AlignmentI cdnaAlignment) | |
| 305 | { | |
| 306 | 4 | if (proteinAlignment == null || cdnaAlignment == null) |
| 307 | { | |
| 308 | 0 | return false; |
| 309 | } | |
| 310 | ||
| 311 | 4 | Set<SequenceI> mappedDna = new HashSet<>(); |
| 312 | 4 | Set<SequenceI> mappedProtein = new HashSet<>(); |
| 313 | ||
| 314 | /* | |
| 315 | * First pass - map sequences where cross-references exist. This include | |
| 316 | * 1-to-many mappings to support, for example, variant cDNA. | |
| 317 | */ | |
| 318 | 4 | boolean mappingPerformed = mapProteinToCdna(proteinAlignment, |
| 319 | cdnaAlignment, mappedDna, mappedProtein, true); | |
| 320 | ||
| 321 | /* | |
| 322 | * Second pass - map sequences where no cross-references exist. This only | |
| 323 | * does 1-to-1 mappings and assumes corresponding sequences are in the same | |
| 324 | * order in the alignments. | |
| 325 | */ | |
| 326 | 4 | mappingPerformed |= mapProteinToCdna(proteinAlignment, cdnaAlignment, |
| 327 | mappedDna, mappedProtein, false); | |
| 328 | 4 | return mappingPerformed; |
| 329 | } | |
| 330 | ||
| 331 | /** | |
| 332 | * Make mappings between compatible sequences (where the cDNA translation | |
| 333 | * matches the protein). | |
| 334 | * | |
| 335 | * @param proteinAlignment | |
| 336 | * @param cdnaAlignment | |
| 337 | * @param mappedDna | |
| 338 | * a set of mapped DNA sequences (to add to) | |
| 339 | * @param mappedProtein | |
| 340 | * a set of mapped Protein sequences (to add to) | |
| 341 | * @param xrefsOnly | |
| 342 | * if true, only map sequences where xrefs exist | |
| 343 | * @return | |
| 344 | */ | |
| 345 | 8 | protected static boolean mapProteinToCdna( |
| 346 | final AlignmentI proteinAlignment, final AlignmentI cdnaAlignment, | |
| 347 | Set<SequenceI> mappedDna, Set<SequenceI> mappedProtein, | |
| 348 | boolean xrefsOnly) | |
| 349 | { | |
| 350 | 8 | boolean mappingExistsOrAdded = false; |
| 351 | 8 | List<SequenceI> thisSeqs = proteinAlignment.getSequences(); |
| 352 | 8 | for (SequenceI aaSeq : thisSeqs) |
| 353 | { | |
| 354 | 22 | boolean proteinMapped = false; |
| 355 | 22 | AlignedCodonFrame acf = new AlignedCodonFrame(); |
| 356 | ||
| 357 | 22 | for (SequenceI cdnaSeq : cdnaAlignment.getSequences()) |
| 358 | { | |
| 359 | /* | |
| 360 | * Always try to map if sequences have xref to each other; this supports | |
| 361 | * variant cDNA or alternative splicing for a protein sequence. | |
| 362 | * | |
| 363 | * If no xrefs, try to map progressively, assuming that alignments have | |
| 364 | * mappable sequences in corresponding order. These are not | |
| 365 | * many-to-many, as that would risk mixing species with similar cDNA | |
| 366 | * sequences. | |
| 367 | */ | |
| 368 | 86 | if (xrefsOnly && !AlignmentUtils.haveCrossRef(aaSeq, cdnaSeq)) |
| 369 | { | |
| 370 | 39 | continue; |
| 371 | } | |
| 372 | ||
| 373 | /* | |
| 374 | * Don't map non-xrefd sequences more than once each. This heuristic | |
| 375 | * allows us to pair up similar sequences in ordered alignments. | |
| 376 | */ | |
| 377 | 47 | if (!xrefsOnly && (mappedProtein.contains(aaSeq) |
| 378 | || mappedDna.contains(cdnaSeq))) | |
| 379 | { | |
| 380 | 29 | continue; |
| 381 | } | |
| 382 | 18 | if (mappingExists(proteinAlignment.getCodonFrames(), |
| 383 | aaSeq.getDatasetSequence(), cdnaSeq.getDatasetSequence())) | |
| 384 | { | |
| 385 | 0 | mappingExistsOrAdded = true; |
| 386 | } | |
| 387 | else | |
| 388 | { | |
| 389 | 18 | MapList map = mapCdnaToProtein(aaSeq, cdnaSeq); |
| 390 | 18 | if (map != null) |
| 391 | { | |
| 392 | 12 | acf.addMap(cdnaSeq, aaSeq, map); |
| 393 | 12 | mappingExistsOrAdded = true; |
| 394 | 12 | proteinMapped = true; |
| 395 | 12 | mappedDna.add(cdnaSeq); |
| 396 | 12 | mappedProtein.add(aaSeq); |
| 397 | } | |
| 398 | } | |
| 399 | } | |
| 400 | 22 | if (proteinMapped) |
| 401 | { | |
| 402 | 11 | proteinAlignment.addCodonFrame(acf); |
| 403 | } | |
| 404 | } | |
| 405 | 8 | return mappingExistsOrAdded; |
| 406 | } | |
| 407 | ||
| 408 | /** | |
| 409 | * Answers true if the mappings include one between the given (dataset) | |
| 410 | * sequences. | |
| 411 | */ | |
| 412 | 18 | protected static boolean mappingExists(List<AlignedCodonFrame> mappings, |
| 413 | SequenceI aaSeq, SequenceI cdnaSeq) | |
| 414 | { | |
| 415 | 18 | if (mappings != null) |
| 416 | { | |
| 417 | 18 | for (AlignedCodonFrame acf : mappings) |
| 418 | { | |
| 419 | 14 | if (cdnaSeq == acf.getDnaForAaSeq(aaSeq)) |
| 420 | { | |
| 421 | 0 | return true; |
| 422 | } | |
| 423 | } | |
| 424 | } | |
| 425 | 18 | return false; |
| 426 | } | |
| 427 | ||
| 428 | /** | |
| 429 | * Builds a mapping (if possible) of a cDNA to a protein sequence. | |
| 430 | * <ul> | |
| 431 | * <li>first checks if the cdna translates exactly to the protein | |
| 432 | * sequence</li> | |
| 433 | * <li>else checks for translation after removing a STOP codon</li> | |
| 434 | * <li>else checks for translation after removing a START codon</li> | |
| 435 | * <li>if that fails, inspect CDS features on the cDNA sequence</li> | |
| 436 | * </ul> | |
| 437 | * Returns null if no mapping is determined. | |
| 438 | * | |
| 439 | * @param proteinSeq | |
| 440 | * the aligned protein sequence | |
| 441 | * @param cdnaSeq | |
| 442 | * the aligned cdna sequence | |
| 443 | * @return | |
| 444 | */ | |
| 445 | 31 | public static MapList mapCdnaToProtein(SequenceI proteinSeq, |
| 446 | SequenceI cdnaSeq) | |
| 447 | { | |
| 448 | /* | |
| 449 | * Here we handle either dataset sequence set (desktop) or absent (applet). | |
| 450 | * Use only the char[] form of the sequence to avoid creating possibly large | |
| 451 | * String objects. | |
| 452 | */ | |
| 453 | 31 | final SequenceI proteinDataset = proteinSeq.getDatasetSequence(); |
| 454 | 31 | char[] aaSeqChars = proteinDataset != null |
| 455 | ? proteinDataset.getSequence() | |
| 456 | : proteinSeq.getSequence(); | |
| 457 | 31 | final SequenceI cdnaDataset = cdnaSeq.getDatasetSequence(); |
| 458 | 31 | char[] cdnaSeqChars = cdnaDataset != null ? cdnaDataset.getSequence() |
| 459 | : cdnaSeq.getSequence(); | |
| 460 | 31 | if (aaSeqChars == null || cdnaSeqChars == null) |
| 461 | { | |
| 462 | 0 | return null; |
| 463 | } | |
| 464 | ||
| 465 | /* | |
| 466 | * cdnaStart/End, proteinStartEnd are base 1 (for dataset sequence mapping) | |
| 467 | */ | |
| 468 | 31 | final int mappedLength = CODON_LENGTH * aaSeqChars.length; |
| 469 | 31 | int cdnaLength = cdnaSeqChars.length; |
| 470 | 31 | int cdnaStart = cdnaSeq.getStart(); |
| 471 | 31 | int cdnaEnd = cdnaSeq.getEnd(); |
| 472 | 31 | final int proteinStart = proteinSeq.getStart(); |
| 473 | 31 | final int proteinEnd = proteinSeq.getEnd(); |
| 474 | ||
| 475 | /* | |
| 476 | * If lengths don't match, try ignoring stop codon (if present) | |
| 477 | */ | |
| 478 | 31 | if (cdnaLength != mappedLength && cdnaLength > 2) |
| 479 | { | |
| 480 | 16 | String lastCodon = String.valueOf(cdnaSeqChars, |
| 481 | cdnaLength - CODON_LENGTH, CODON_LENGTH) | |
| 482 | .toUpperCase(Locale.ROOT); | |
| 483 | 16 | for (String stop : ResidueProperties.STOP_CODONS) |
| 484 | { | |
| 485 | 47 | if (lastCodon.equals(stop)) |
| 486 | { | |
| 487 | 2 | cdnaEnd -= CODON_LENGTH; |
| 488 | 2 | cdnaLength -= CODON_LENGTH; |
| 489 | 2 | break; |
| 490 | } | |
| 491 | } | |
| 492 | } | |
| 493 | ||
| 494 | /* | |
| 495 | * If lengths still don't match, try ignoring start codon. | |
| 496 | */ | |
| 497 | 31 | int startOffset = 0; |
| 498 | 31 | if (cdnaLength != mappedLength && cdnaLength > 2 |
| 499 | && String.valueOf(cdnaSeqChars, 0, CODON_LENGTH) | |
| 500 | .toUpperCase(Locale.ROOT) | |
| 501 | .equals(ResidueProperties.START)) | |
| 502 | { | |
| 503 | 5 | startOffset += CODON_LENGTH; |
| 504 | 5 | cdnaStart += CODON_LENGTH; |
| 505 | 5 | cdnaLength -= CODON_LENGTH; |
| 506 | } | |
| 507 | ||
| 508 | 31 | if (translatesAs(cdnaSeqChars, startOffset, aaSeqChars)) |
| 509 | { | |
| 510 | /* | |
| 511 | * protein is translation of dna (+/- start/stop codons) | |
| 512 | */ | |
| 513 | 15 | MapList map = new MapList(new int[] { cdnaStart, cdnaEnd }, |
| 514 | new int[] | |
| 515 | { proteinStart, proteinEnd }, CODON_LENGTH, 1); | |
| 516 | 15 | return map; |
| 517 | } | |
| 518 | ||
| 519 | /* | |
| 520 | * translation failed - try mapping CDS annotated regions of dna | |
| 521 | */ | |
| 522 | 16 | return mapCdsToProtein(cdnaSeq, proteinSeq); |
| 523 | } | |
| 524 | ||
| 525 | /** | |
| 526 | * Test whether the given cdna sequence, starting at the given offset, | |
| 527 | * translates to the given amino acid sequence, using the standard translation | |
| 528 | * table. Designed to fail fast i.e. as soon as a mismatch position is found. | |
| 529 | * | |
| 530 | * @param cdnaSeqChars | |
| 531 | * @param cdnaStart | |
| 532 | * @param aaSeqChars | |
| 533 | * @return | |
| 534 | */ | |
| 535 | 51 | protected static boolean translatesAs(char[] cdnaSeqChars, int cdnaStart, |
| 536 | char[] aaSeqChars) | |
| 537 | { | |
| 538 | 51 | if (cdnaSeqChars == null || aaSeqChars == null) |
| 539 | { | |
| 540 | 3 | return false; |
| 541 | } | |
| 542 | ||
| 543 | 48 | int aaPos = 0; |
| 544 | 48 | int dnaPos = cdnaStart; |
| 545 | 161 | for (; dnaPos < cdnaSeqChars.length - 2 |
| 546 | && aaPos < aaSeqChars.length; dnaPos += CODON_LENGTH, aaPos++) | |
| 547 | { | |
| 548 | 130 | String codon = String.valueOf(cdnaSeqChars, dnaPos, CODON_LENGTH); |
| 549 | 130 | final String translated = ResidueProperties.codonTranslate(codon); |
| 550 | ||
| 551 | /* | |
| 552 | * allow * in protein to match untranslatable in dna | |
| 553 | */ | |
| 554 | 130 | final char aaRes = aaSeqChars[aaPos]; |
| 555 | 130 | if ((translated == null || ResidueProperties.STOP.equals(translated)) |
| 556 | && aaRes == '*') | |
| 557 | { | |
| 558 | 4 | continue; |
| 559 | } | |
| 560 | 126 | if (translated == null || !(aaRes == translated.charAt(0))) |
| 561 | { | |
| 562 | // debug | |
| 563 | // jalview.bin.Console.outPrintln(("Mismatch at " + i + "/" + aaResidue | |
| 564 | // + ": " | |
| 565 | // + codon + "(" + translated + ") != " + aaRes)); | |
| 566 | 17 | return false; |
| 567 | } | |
| 568 | } | |
| 569 | ||
| 570 | /* | |
| 571 | * check we matched all of the protein sequence | |
| 572 | */ | |
| 573 | 31 | if (aaPos != aaSeqChars.length) |
| 574 | { | |
| 575 | 2 | return false; |
| 576 | } | |
| 577 | ||
| 578 | /* | |
| 579 | * check we matched all of the dna except | |
| 580 | * for optional trailing STOP codon | |
| 581 | */ | |
| 582 | 29 | if (dnaPos == cdnaSeqChars.length) |
| 583 | { | |
| 584 | 17 | return true; |
| 585 | } | |
| 586 | 12 | if (dnaPos == cdnaSeqChars.length - CODON_LENGTH) |
| 587 | { | |
| 588 | 11 | String codon = String.valueOf(cdnaSeqChars, dnaPos, CODON_LENGTH); |
| 589 | 11 | if (ResidueProperties.STOP |
| 590 | .equals(ResidueProperties.codonTranslate(codon))) | |
| 591 | { | |
| 592 | 9 | return true; |
| 593 | } | |
| 594 | } | |
| 595 | 3 | return false; |
| 596 | } | |
| 597 | ||
| 598 | /** | |
| 599 | * Align sequence 'seq' to match the alignment of a mapped sequence. Note this | |
| 600 | * currently assumes that we are aligning cDNA to match protein. | |
| 601 | * | |
| 602 | * @param seq | |
| 603 | * the sequence to be realigned | |
| 604 | * @param al | |
| 605 | * the alignment whose sequence alignment is to be 'copied' | |
| 606 | * @param gap | |
| 607 | * character string represent a gap in the realigned sequence | |
| 608 | * @param preserveUnmappedGaps | |
| 609 | * @param preserveMappedGaps | |
| 610 | * @return true if the sequence was realigned, false if it could not be | |
| 611 | */ | |
| 612 | 8 | public static boolean alignSequenceAs(SequenceI seq, AlignmentI al, |
| 613 | String gap, boolean preserveMappedGaps, | |
| 614 | boolean preserveUnmappedGaps) | |
| 615 | { | |
| 616 | /* | |
| 617 | * Get any mappings from the source alignment to the target (dataset) | |
| 618 | * sequence. | |
| 619 | */ | |
| 620 | // TODO there may be one AlignedCodonFrame per dataset sequence, or one with | |
| 621 | // all mappings. Would it help to constrain this? | |
| 622 | 8 | List<AlignedCodonFrame> mappings = al.getCodonFrame(seq); |
| 623 | 8 | if (mappings == null || mappings.isEmpty()) |
| 624 | { | |
| 625 | 0 | return false; |
| 626 | } | |
| 627 | ||
| 628 | /* | |
| 629 | * Locate the aligned source sequence whose dataset sequence is mapped. We | |
| 630 | * just take the first match here (as we can't align like more than one | |
| 631 | * sequence). | |
| 632 | */ | |
| 633 | 8 | SequenceI alignFrom = null; |
| 634 | 8 | AlignedCodonFrame mapping = null; |
| 635 | 8 | for (AlignedCodonFrame mp : mappings) |
| 636 | { | |
| 637 | 8 | alignFrom = mp.findAlignedSequence(seq, al); |
| 638 | 8 | if (alignFrom != null) |
| 639 | { | |
| 640 | 4 | mapping = mp; |
| 641 | 4 | break; |
| 642 | } | |
| 643 | } | |
| 644 | ||
| 645 | 8 | if (alignFrom == null) |
| 646 | { | |
| 647 | 4 | return false; |
| 648 | } | |
| 649 | 4 | alignSequenceAs(seq, alignFrom, mapping, gap, al.getGapCharacter(), |
| 650 | preserveMappedGaps, preserveUnmappedGaps); | |
| 651 | 4 | return true; |
| 652 | } | |
| 653 | ||
| 654 | /** | |
| 655 | * Align sequence 'alignTo' the same way as 'alignFrom', using the mapping to | |
| 656 | * match residues and codons. Flags control whether existing gaps in unmapped | |
| 657 | * (intron) and mapped (exon) regions are preserved or not. Gaps between | |
| 658 | * intron and exon are only retained if both flags are set. | |
| 659 | * | |
| 660 | * @param alignTo | |
| 661 | * @param alignFrom | |
| 662 | * @param mapping | |
| 663 | * @param myGap | |
| 664 | * @param sourceGap | |
| 665 | * @param preserveUnmappedGaps | |
| 666 | * @param preserveMappedGaps | |
| 667 | */ | |
| 668 | 19 | public static void alignSequenceAs(SequenceI alignTo, SequenceI alignFrom, |
| 669 | AlignedCodonFrame mapping, String myGap, char sourceGap, | |
| 670 | boolean preserveMappedGaps, boolean preserveUnmappedGaps) | |
| 671 | { | |
| 672 | // TODO generalise to work for Protein-Protein, dna-dna, dna-protein | |
| 673 | ||
| 674 | // aligned and dataset sequence positions, all base zero | |
| 675 | 19 | int thisSeqPos = 0; |
| 676 | 19 | int sourceDsPos = 0; |
| 677 | ||
| 678 | 19 | int basesWritten = 0; |
| 679 | 19 | char myGapChar = myGap.charAt(0); |
| 680 | 19 | int ratio = myGap.length(); |
| 681 | ||
| 682 | 19 | int fromOffset = alignFrom.getStart() - 1; |
| 683 | 19 | int toOffset = alignTo.getStart() - 1; |
| 684 | 19 | int sourceGapMappedLength = 0; |
| 685 | 19 | boolean inExon = false; |
| 686 | 19 | final int toLength = alignTo.getLength(); |
| 687 | 19 | final int fromLength = alignFrom.getLength(); |
| 688 | 19 | StringBuilder thisAligned = new StringBuilder(2 * toLength); |
| 689 | ||
| 690 | /* | |
| 691 | * Traverse the 'model' aligned sequence | |
| 692 | */ | |
| 693 | 205 | for (int i = 0; i < fromLength; i++) |
| 694 | { | |
| 695 | 186 | char sourceChar = alignFrom.getCharAt(i); |
| 696 | 186 | if (sourceChar == sourceGap) |
| 697 | { | |
| 698 | 44 | sourceGapMappedLength += ratio; |
| 699 | 44 | continue; |
| 700 | } | |
| 701 | ||
| 702 | /* | |
| 703 | * Found a non-gap character. Locate its mapped region if any. | |
| 704 | */ | |
| 705 | 142 | sourceDsPos++; |
| 706 | // Note mapping positions are base 1, our sequence positions base 0 | |
| 707 | 142 | int[] mappedPos = mapping.getMappedRegion(alignTo, alignFrom, |
| 708 | sourceDsPos + fromOffset); | |
| 709 | 142 | if (mappedPos == null) |
| 710 | { | |
| 711 | /* | |
| 712 | * unmapped position; treat like a gap | |
| 713 | */ | |
| 714 | 94 | sourceGapMappedLength += ratio; |
| 715 | // jalview.bin.Console.errPrintln("Can't align: no codon mapping to | |
| 716 | // residue " | |
| 717 | // + sourceDsPos + "(" + sourceChar + ")"); | |
| 718 | // return; | |
| 719 | 94 | continue; |
| 720 | } | |
| 721 | ||
| 722 | 48 | int mappedCodonStart = mappedPos[0]; // position (1...) of codon start |
| 723 | 48 | int mappedCodonEnd = mappedPos[mappedPos.length - 1]; // codon end pos |
| 724 | 48 | StringBuilder trailingCopiedGap = new StringBuilder(); |
| 725 | ||
| 726 | /* | |
| 727 | * Copy dna sequence up to and including this codon. Optionally, include | |
| 728 | * gaps before the codon starts (in introns) and/or after the codon starts | |
| 729 | * (in exons). | |
| 730 | * | |
| 731 | * Note this only works for 'linear' splicing, not reverse or interleaved. | |
| 732 | * But then 'align dna as protein' doesn't make much sense otherwise. | |
| 733 | */ | |
| 734 | 48 | int intronLength = 0; |
| 735 | 294 | while (basesWritten + toOffset < mappedCodonEnd |
| 736 | && thisSeqPos < toLength) | |
| 737 | { | |
| 738 | 246 | final char c = alignTo.getCharAt(thisSeqPos++); |
| 739 | 246 | if (c != myGapChar) |
| 740 | { | |
| 741 | 146 | basesWritten++; |
| 742 | 146 | int sourcePosition = basesWritten + toOffset; |
| 743 | 146 | if (sourcePosition < mappedCodonStart) |
| 744 | { | |
| 745 | /* | |
| 746 | * Found an unmapped (intron) base. First add in any preceding gaps | |
| 747 | * (if wanted). | |
| 748 | */ | |
| 749 | 48 | if (preserveUnmappedGaps && trailingCopiedGap.length() > 0) |
| 750 | { | |
| 751 | 17 | thisAligned.append(trailingCopiedGap.toString()); |
| 752 | 17 | intronLength += trailingCopiedGap.length(); |
| 753 | 17 | trailingCopiedGap = new StringBuilder(); |
| 754 | } | |
| 755 | 48 | intronLength++; |
| 756 | 48 | inExon = false; |
| 757 | } | |
| 758 | else | |
| 759 | { | |
| 760 | 98 | final boolean startOfCodon = sourcePosition == mappedCodonStart; |
| 761 | 98 | int gapsToAdd = calculateGapsToInsert(preserveMappedGaps, |
| 762 | preserveUnmappedGaps, sourceGapMappedLength, inExon, | |
| 763 | trailingCopiedGap.length(), intronLength, startOfCodon); | |
| 764 | 215 | for (int k = 0; k < gapsToAdd; k++) |
| 765 | { | |
| 766 | 117 | thisAligned.append(myGapChar); |
| 767 | } | |
| 768 | 98 | sourceGapMappedLength = 0; |
| 769 | 98 | inExon = true; |
| 770 | } | |
| 771 | 146 | thisAligned.append(c); |
| 772 | 146 | trailingCopiedGap = new StringBuilder(); |
| 773 | } | |
| 774 | else | |
| 775 | { | |
| 776 | 100 | if (inExon && preserveMappedGaps) |
| 777 | { | |
| 778 | 32 | trailingCopiedGap.append(myGapChar); |
| 779 | } | |
| 780 | 68 | else if (!inExon && preserveUnmappedGaps) |
| 781 | { | |
| 782 | 27 | trailingCopiedGap.append(myGapChar); |
| 783 | } | |
| 784 | } | |
| 785 | } | |
| 786 | } | |
| 787 | ||
| 788 | /* | |
| 789 | * At end of model aligned sequence. Copy any remaining target sequence, optionally | |
| 790 | * including (intron) gaps. | |
| 791 | */ | |
| 792 | 129 | while (thisSeqPos < toLength) |
| 793 | { | |
| 794 | 110 | final char c = alignTo.getCharAt(thisSeqPos++); |
| 795 | 110 | if (c != myGapChar || preserveUnmappedGaps) |
| 796 | { | |
| 797 | 102 | thisAligned.append(c); |
| 798 | } | |
| 799 | 110 | sourceGapMappedLength--; |
| 800 | } | |
| 801 | ||
| 802 | /* | |
| 803 | * finally add gaps to pad for any trailing source gaps or | |
| 804 | * unmapped characters | |
| 805 | */ | |
| 806 | 19 | if (preserveUnmappedGaps) |
| 807 | { | |
| 808 | 24 | while (sourceGapMappedLength > 0) |
| 809 | { | |
| 810 | 12 | thisAligned.append(myGapChar); |
| 811 | 12 | sourceGapMappedLength--; |
| 812 | } | |
| 813 | } | |
| 814 | ||
| 815 | /* | |
| 816 | * All done aligning, set the aligned sequence. | |
| 817 | */ | |
| 818 | 19 | alignTo.setSequence(new String(thisAligned)); |
| 819 | } | |
| 820 | ||
| 821 | /** | |
| 822 | * Helper method to work out how many gaps to insert when realigning. | |
| 823 | * | |
| 824 | * @param preserveMappedGaps | |
| 825 | * @param preserveUnmappedGaps | |
| 826 | * @param sourceGapMappedLength | |
| 827 | * @param inExon | |
| 828 | * @param trailingCopiedGap | |
| 829 | * @param intronLength | |
| 830 | * @param startOfCodon | |
| 831 | * @return | |
| 832 | */ | |
| 833 | 98 | protected static int calculateGapsToInsert(boolean preserveMappedGaps, |
| 834 | boolean preserveUnmappedGaps, int sourceGapMappedLength, | |
| 835 | boolean inExon, int trailingGapLength, int intronLength, | |
| 836 | final boolean startOfCodon) | |
| 837 | { | |
| 838 | 98 | int gapsToAdd = 0; |
| 839 | 98 | if (startOfCodon) |
| 840 | { | |
| 841 | /* | |
| 842 | * Reached start of codon. Ignore trailing gaps in intron unless we are | |
| 843 | * preserving gaps in both exon and intron. Ignore them anyway if the | |
| 844 | * protein alignment introduces a gap at least as large as the intronic | |
| 845 | * region. | |
| 846 | */ | |
| 847 | 40 | if (inExon && !preserveMappedGaps) |
| 848 | { | |
| 849 | 4 | trailingGapLength = 0; |
| 850 | } | |
| 851 | 40 | if (!inExon && !(preserveMappedGaps && preserveUnmappedGaps)) |
| 852 | { | |
| 853 | 19 | trailingGapLength = 0; |
| 854 | } | |
| 855 | 40 | if (inExon) |
| 856 | { | |
| 857 | 14 | gapsToAdd = Math.max(sourceGapMappedLength, trailingGapLength); |
| 858 | } | |
| 859 | else | |
| 860 | { | |
| 861 | 26 | if (intronLength + trailingGapLength <= sourceGapMappedLength) |
| 862 | { | |
| 863 | 20 | gapsToAdd = sourceGapMappedLength - intronLength; |
| 864 | } | |
| 865 | else | |
| 866 | { | |
| 867 | 6 | gapsToAdd = Math.min( |
| 868 | intronLength + trailingGapLength - sourceGapMappedLength, | |
| 869 | trailingGapLength); | |
| 870 | } | |
| 871 | } | |
| 872 | } | |
| 873 | else | |
| 874 | { | |
| 875 | /* | |
| 876 | * second or third base of codon; check for any gaps in dna | |
| 877 | */ | |
| 878 | 58 | if (!preserveMappedGaps) |
| 879 | { | |
| 880 | 32 | trailingGapLength = 0; |
| 881 | } | |
| 882 | 58 | gapsToAdd = Math.max(sourceGapMappedLength, trailingGapLength); |
| 883 | } | |
| 884 | 98 | return gapsToAdd; |
| 885 | } | |
| 886 | ||
| 887 | /** | |
| 888 | * Realigns the given protein to match the alignment of the dna, using codon | |
| 889 | * mappings to translate aligned codon positions to protein residues. | |
| 890 | * | |
| 891 | * @param protein | |
| 892 | * the alignment whose sequences are realigned by this method | |
| 893 | * @param dna | |
| 894 | * the dna alignment whose alignment we are 'copying' | |
| 895 | * @return the number of sequences that were realigned | |
| 896 | */ | |
| 897 | 5 | public static int alignProteinAsDna(AlignmentI protein, AlignmentI dna) |
| 898 | { | |
| 899 | 5 | if (protein.isNucleotide() || !dna.isNucleotide()) |
| 900 | { | |
| 901 | 0 | jalview.bin.Console |
| 902 | .errPrintln("Wrong alignment type in alignProteinAsDna"); | |
| 903 | 0 | return 0; |
| 904 | } | |
| 905 | 5 | List<SequenceI> unmappedProtein = new ArrayList<>(); |
| 906 | 5 | Map<AlignedCodon, Map<SequenceI, AlignedCodon>> alignedCodons = buildCodonColumnsMap( |
| 907 | protein, dna, unmappedProtein); | |
| 908 | 5 | return alignProteinAs(protein, alignedCodons, unmappedProtein); |
| 909 | } | |
| 910 | ||
| 911 | /** | |
| 912 | * Realigns the given dna to match the alignment of the protein, using codon | |
| 913 | * mappings to translate aligned peptide positions to codons. | |
| 914 | * | |
| 915 | * Always produces a padded CDS alignment. | |
| 916 | * | |
| 917 | * @param dna | |
| 918 | * the alignment whose sequences are realigned by this method | |
| 919 | * @param protein | |
| 920 | * the protein alignment whose alignment we are 'copying' | |
| 921 | * @return the number of sequences that were realigned | |
| 922 | */ | |
| 923 | 4 | public static int alignCdsAsProtein(AlignmentI dna, AlignmentI protein) |
| 924 | { | |
| 925 | 4 | if (protein.isNucleotide() || !dna.isNucleotide()) |
| 926 | { | |
| 927 | 0 | jalview.bin.Console |
| 928 | .errPrintln("Wrong alignment type in alignProteinAsDna"); | |
| 929 | 0 | return 0; |
| 930 | } | |
| 931 | // todo: implement this | |
| 932 | 4 | List<AlignedCodonFrame> mappings = protein.getCodonFrames(); |
| 933 | 4 | int alignedCount = 0; |
| 934 | 4 | int width = 0; // alignment width for padding CDS |
| 935 | 4 | for (SequenceI dnaSeq : dna.getSequences()) |
| 936 | { | |
| 937 | 5 | if (alignCdsSequenceAsProtein(dnaSeq, protein, mappings, |
| 938 | dna.getGapCharacter())) | |
| 939 | { | |
| 940 | 5 | alignedCount++; |
| 941 | } | |
| 942 | 5 | width = Math.max(dnaSeq.getLength(), width); |
| 943 | } | |
| 944 | 4 | int oldwidth; |
| 945 | 4 | int diff; |
| 946 | 4 | for (SequenceI dnaSeq : dna.getSequences()) |
| 947 | { | |
| 948 | 5 | oldwidth = dnaSeq.getLength(); |
| 949 | 5 | diff = width - oldwidth; |
| 950 | 5 | if (diff > 0) |
| 951 | { | |
| 952 | 1 | dnaSeq.insertCharAt(oldwidth, diff, dna.getGapCharacter()); |
| 953 | } | |
| 954 | } | |
| 955 | 4 | return alignedCount; |
| 956 | } | |
| 957 | ||
| 958 | /** | |
| 959 | * Helper method to align (if possible) the dna sequence to match the | |
| 960 | * alignment of a mapped protein sequence. This is currently limited to | |
| 961 | * handling coding sequence only. | |
| 962 | * | |
| 963 | * @param cdsSeq | |
| 964 | * @param protein | |
| 965 | * @param mappings | |
| 966 | * @param gapChar | |
| 967 | * @return | |
| 968 | */ | |
| 969 | 5 | static boolean alignCdsSequenceAsProtein(SequenceI cdsSeq, |
| 970 | AlignmentI protein, List<AlignedCodonFrame> mappings, | |
| 971 | char gapChar) | |
| 972 | { | |
| 973 | 5 | SequenceI cdsDss = cdsSeq.getDatasetSequence(); |
| 974 | 5 | if (cdsDss == null) |
| 975 | { | |
| 976 | 0 | System.err |
| 977 | .println("alignCdsSequenceAsProtein needs aligned sequence!"); | |
| 978 | 0 | return false; |
| 979 | } | |
| 980 | ||
| 981 | 5 | List<AlignedCodonFrame> dnaMappings = MappingUtils |
| 982 | .findMappingsForSequence(cdsSeq, mappings); | |
| 983 | 5 | for (AlignedCodonFrame mapping : dnaMappings) |
| 984 | { | |
| 985 | 5 | SequenceI peptide = mapping.findAlignedSequence(cdsSeq, protein); |
| 986 | 5 | if (peptide != null) |
| 987 | { | |
| 988 | 5 | final int peptideLength = peptide.getLength(); |
| 989 | 5 | Mapping map = mapping.getMappingBetween(cdsSeq, peptide); |
| 990 | 5 | if (map != null) |
| 991 | { | |
| 992 | 5 | MapList mapList = map.getMap(); |
| 993 | 5 | if (map.getTo() == peptide.getDatasetSequence()) |
| 994 | { | |
| 995 | 5 | mapList = mapList.getInverse(); |
| 996 | } | |
| 997 | 5 | final int cdsLength = cdsDss.getLength(); |
| 998 | 5 | int mappedFromLength = MappingUtils |
| 999 | .getLength(mapList.getFromRanges()); | |
| 1000 | 5 | int mappedToLength = MappingUtils |
| 1001 | .getLength(mapList.getToRanges()); | |
| 1002 | 5 | boolean addStopCodon = (cdsLength == mappedFromLength |
| 1003 | * CODON_LENGTH + CODON_LENGTH) | |
| 1004 | || (peptide.getDatasetSequence() | |
| 1005 | .getLength() == mappedFromLength - 1); | |
| 1006 | 5 | if (cdsLength != mappedToLength && !addStopCodon) |
| 1007 | { | |
| 1008 | 0 | jalview.bin.Console.errPrintln(String.format( |
| 1009 | "Can't align cds as protein (length mismatch %d/%d): %s", | |
| 1010 | cdsLength, mappedToLength, cdsSeq.getName())); | |
| 1011 | } | |
| 1012 | ||
| 1013 | /* | |
| 1014 | * pre-fill the aligned cds sequence with gaps | |
| 1015 | */ | |
| 1016 | 5 | char[] alignedCds = new char[peptideLength * CODON_LENGTH |
| 1017 | 5 | + (addStopCodon ? CODON_LENGTH : 0)]; |
| 1018 | 5 | Arrays.fill(alignedCds, gapChar); |
| 1019 | ||
| 1020 | /* | |
| 1021 | * walk over the aligned peptide sequence and insert mapped | |
| 1022 | * codons for residues in the aligned cds sequence | |
| 1023 | */ | |
| 1024 | 5 | int copiedBases = 0; |
| 1025 | 5 | int cdsStart = cdsDss.getStart(); |
| 1026 | 5 | int proteinPos = peptide.getStart() - 1; |
| 1027 | 5 | int cdsCol = 0; |
| 1028 | ||
| 1029 | 33 | for (int col = 0; col < peptideLength; col++) |
| 1030 | { | |
| 1031 | 28 | char residue = peptide.getCharAt(col); |
| 1032 | ||
| 1033 | 28 | if (Comparison.isGap(residue)) |
| 1034 | { | |
| 1035 | 13 | cdsCol += CODON_LENGTH; |
| 1036 | } | |
| 1037 | else | |
| 1038 | { | |
| 1039 | 15 | proteinPos++; |
| 1040 | 15 | int[] codon = mapList.locateInTo(proteinPos, proteinPos); |
| 1041 | 15 | if (codon == null) |
| 1042 | { | |
| 1043 | // e.g. incomplete start codon, X in peptide | |
| 1044 | 0 | cdsCol += CODON_LENGTH; |
| 1045 | } | |
| 1046 | else | |
| 1047 | { | |
| 1048 | 60 | for (int j = codon[0]; j <= codon[1]; j++) |
| 1049 | { | |
| 1050 | 45 | char mappedBase = cdsDss.getCharAt(j - cdsStart); |
| 1051 | 45 | alignedCds[cdsCol++] = mappedBase; |
| 1052 | 45 | copiedBases++; |
| 1053 | } | |
| 1054 | } | |
| 1055 | } | |
| 1056 | } | |
| 1057 | ||
| 1058 | /* | |
| 1059 | * append stop codon if not mapped from protein, | |
| 1060 | * closing it up to the end of the mapped sequence | |
| 1061 | */ | |
| 1062 | 5 | if (copiedBases == cdsLength - CODON_LENGTH) |
| 1063 | { | |
| 1064 | 0 | for (int i = alignedCds.length - 1; i >= 0; i--) |
| 1065 | { | |
| 1066 | 0 | if (!Comparison.isGap(alignedCds[i])) |
| 1067 | { | |
| 1068 | 0 | cdsCol = i + 1; // gap just after end of sequence |
| 1069 | 0 | break; |
| 1070 | } | |
| 1071 | } | |
| 1072 | 0 | for (int i = cdsLength - CODON_LENGTH; i < cdsLength; i++) |
| 1073 | { | |
| 1074 | 0 | alignedCds[cdsCol++] = cdsDss.getCharAt(i); |
| 1075 | } | |
| 1076 | } | |
| 1077 | 5 | cdsSeq.setSequence(new String(alignedCds)); |
| 1078 | 5 | return true; |
| 1079 | } | |
| 1080 | } | |
| 1081 | } | |
| 1082 | 0 | return false; |
| 1083 | } | |
| 1084 | ||
| 1085 | /** | |
| 1086 | * Builds a map whose key is an aligned codon position (3 alignment column | |
| 1087 | * numbers base 0), and whose value is a map from protein sequence to each | |
| 1088 | * protein's peptide residue for that codon. The map generates an ordering of | |
| 1089 | * the codons, and allows us to read off the peptides at each position in | |
| 1090 | * order to assemble 'aligned' protein sequences. | |
| 1091 | * | |
| 1092 | * @param protein | |
| 1093 | * the protein alignment | |
| 1094 | * @param dna | |
| 1095 | * the coding dna alignment | |
| 1096 | * @param unmappedProtein | |
| 1097 | * any unmapped proteins are added to this list | |
| 1098 | * @return | |
| 1099 | */ | |
| 1100 | 5 | protected static Map<AlignedCodon, Map<SequenceI, AlignedCodon>> buildCodonColumnsMap( |
| 1101 | AlignmentI protein, AlignmentI dna, | |
| 1102 | List<SequenceI> unmappedProtein) | |
| 1103 | { | |
| 1104 | /* | |
| 1105 | * maintain a list of any proteins with no mappings - these will be | |
| 1106 | * rendered 'as is' in the protein alignment as we can't align them | |
| 1107 | */ | |
| 1108 | 5 | unmappedProtein.addAll(protein.getSequences()); |
| 1109 | ||
| 1110 | 5 | List<AlignedCodonFrame> mappings = protein.getCodonFrames(); |
| 1111 | ||
| 1112 | /* | |
| 1113 | * Map will hold, for each aligned codon position e.g. [3, 5, 6], a map of | |
| 1114 | * {dnaSequence, {proteinSequence, codonProduct}} at that position. The | |
| 1115 | * comparator keeps the codon positions ordered. | |
| 1116 | */ | |
| 1117 | 5 | Map<AlignedCodon, Map<SequenceI, AlignedCodon>> alignedCodons = new TreeMap<>( |
| 1118 | new CodonComparator()); | |
| 1119 | ||
| 1120 | 5 | for (SequenceI dnaSeq : dna.getSequences()) |
| 1121 | { | |
| 1122 | 30 | for (AlignedCodonFrame mapping : mappings) |
| 1123 | { | |
| 1124 | 516 | SequenceI prot = mapping.findAlignedSequence(dnaSeq, protein); |
| 1125 | 516 | if (prot != null) |
| 1126 | { | |
| 1127 | 32 | Mapping seqMap = mapping.getMappingForSequence(dnaSeq); |
| 1128 | 32 | addCodonPositions(dnaSeq, prot, protein.getGapCharacter(), seqMap, |
| 1129 | alignedCodons); | |
| 1130 | 32 | unmappedProtein.remove(prot); |
| 1131 | } | |
| 1132 | } | |
| 1133 | } | |
| 1134 | ||
| 1135 | /* | |
| 1136 | * Finally add any unmapped peptide start residues (e.g. for incomplete | |
| 1137 | * codons) as if at the codon position before the second residue | |
| 1138 | */ | |
| 1139 | // TODO resolve JAL-2022 so this fudge can be removed | |
| 1140 | 5 | int mappedSequenceCount = protein.getHeight() - unmappedProtein.size(); |
| 1141 | 5 | addUnmappedPeptideStarts(alignedCodons, mappedSequenceCount); |
| 1142 | ||
| 1143 | 5 | return alignedCodons; |
| 1144 | } | |
| 1145 | ||
| 1146 | /** | |
| 1147 | * Scans for any protein mapped from position 2 (meaning unmapped start | |
| 1148 | * position e.g. an incomplete codon), and synthesizes a 'codon' for it at the | |
| 1149 | * preceding position in the alignment | |
| 1150 | * | |
| 1151 | * @param alignedCodons | |
| 1152 | * the codon-to-peptide map | |
| 1153 | * @param mappedSequenceCount | |
| 1154 | * the number of distinct sequences in the map | |
| 1155 | */ | |
| 1156 | 5 | protected static void addUnmappedPeptideStarts( |
| 1157 | Map<AlignedCodon, Map<SequenceI, AlignedCodon>> alignedCodons, | |
| 1158 | int mappedSequenceCount) | |
| 1159 | { | |
| 1160 | // TODO delete this ugly hack once JAL-2022 is resolved | |
| 1161 | // i.e. we can model startPhase > 0 (incomplete start codon) | |
| 1162 | ||
| 1163 | 5 | List<SequenceI> sequencesChecked = new ArrayList<>(); |
| 1164 | 5 | AlignedCodon lastCodon = null; |
| 1165 | 5 | Map<SequenceI, AlignedCodon> toAdd = new HashMap<>(); |
| 1166 | ||
| 1167 | 5 | for (Entry<AlignedCodon, Map<SequenceI, AlignedCodon>> entry : alignedCodons |
| 1168 | .entrySet()) | |
| 1169 | { | |
| 1170 | 1913 | for (Entry<SequenceI, AlignedCodon> sequenceCodon : entry.getValue() |
| 1171 | .entrySet()) | |
| 1172 | { | |
| 1173 | 10671 | SequenceI seq = sequenceCodon.getKey(); |
| 1174 | 10671 | if (sequencesChecked.contains(seq)) |
| 1175 | { | |
| 1176 | 10641 | continue; |
| 1177 | } | |
| 1178 | 30 | sequencesChecked.add(seq); |
| 1179 | 30 | AlignedCodon codon = sequenceCodon.getValue(); |
| 1180 | 30 | if (codon.peptideCol > 1) |
| 1181 | { | |
| 1182 | 0 | jalview.bin.Console.errPrintln( |
| 1183 | "Problem mapping protein with >1 unmapped start positions: " | |
| 1184 | + seq.getName()); | |
| 1185 | } | |
| 1186 | 30 | else if (codon.peptideCol == 1) |
| 1187 | { | |
| 1188 | /* | |
| 1189 | * first position (peptideCol == 0) was unmapped - add it | |
| 1190 | */ | |
| 1191 | 6 | if (lastCodon != null) |
| 1192 | { | |
| 1193 | 5 | AlignedCodon firstPeptide = new AlignedCodon(lastCodon.pos1, |
| 1194 | lastCodon.pos2, lastCodon.pos3, | |
| 1195 | String.valueOf(seq.getCharAt(0)), 0); | |
| 1196 | 5 | toAdd.put(seq, firstPeptide); |
| 1197 | } | |
| 1198 | else | |
| 1199 | { | |
| 1200 | /* | |
| 1201 | * unmapped residue at start of alignment (no prior column) - | |
| 1202 | * 'insert' at nominal codon [0, 0, 0] | |
| 1203 | */ | |
| 1204 | 1 | AlignedCodon firstPeptide = new AlignedCodon(0, 0, 0, |
| 1205 | String.valueOf(seq.getCharAt(0)), 0); | |
| 1206 | 1 | toAdd.put(seq, firstPeptide); |
| 1207 | } | |
| 1208 | } | |
| 1209 | 30 | if (sequencesChecked.size() == mappedSequenceCount) |
| 1210 | { | |
| 1211 | // no need to check past first mapped position in all sequences | |
| 1212 | 5 | break; |
| 1213 | } | |
| 1214 | } | |
| 1215 | 1913 | lastCodon = entry.getKey(); |
| 1216 | } | |
| 1217 | ||
| 1218 | /* | |
| 1219 | * add any new codons safely after iterating over the map | |
| 1220 | */ | |
| 1221 | 5 | for (Entry<SequenceI, AlignedCodon> startCodon : toAdd.entrySet()) |
| 1222 | { | |
| 1223 | 6 | addCodonToMap(alignedCodons, startCodon.getValue(), |
| 1224 | startCodon.getKey()); | |
| 1225 | } | |
| 1226 | } | |
| 1227 | ||
| 1228 | /** | |
| 1229 | * Update the aligned protein sequences to match the codon alignments given in | |
| 1230 | * the map. | |
| 1231 | * | |
| 1232 | * @param protein | |
| 1233 | * @param alignedCodons | |
| 1234 | * an ordered map of codon positions (columns), with sequence/peptide | |
| 1235 | * values present in each column | |
| 1236 | * @param unmappedProtein | |
| 1237 | * @return | |
| 1238 | */ | |
| 1239 | 5 | protected static int alignProteinAs(AlignmentI protein, |
| 1240 | Map<AlignedCodon, Map<SequenceI, AlignedCodon>> alignedCodons, | |
| 1241 | List<SequenceI> unmappedProtein) | |
| 1242 | { | |
| 1243 | /* | |
| 1244 | * prefill peptide sequences with gaps | |
| 1245 | */ | |
| 1246 | 5 | int alignedWidth = alignedCodons.size(); |
| 1247 | 5 | char[] gaps = new char[alignedWidth]; |
| 1248 | 5 | Arrays.fill(gaps, protein.getGapCharacter()); |
| 1249 | 5 | Map<SequenceI, char[]> peptides = new HashMap<>(); |
| 1250 | 5 | for (SequenceI seq : protein.getSequences()) |
| 1251 | { | |
| 1252 | 31 | if (!unmappedProtein.contains(seq)) |
| 1253 | { | |
| 1254 | 30 | peptides.put(seq, Arrays.copyOf(gaps, gaps.length)); |
| 1255 | } | |
| 1256 | } | |
| 1257 | ||
| 1258 | /* | |
| 1259 | * Traverse the codons left to right (as defined by CodonComparator) | |
| 1260 | * and insert peptides in each column where the sequence is mapped. | |
| 1261 | * This gives a peptide 'alignment' where residues are aligned if their | |
| 1262 | * corresponding codons occupy the same columns in the cdna alignment. | |
| 1263 | */ | |
| 1264 | 5 | int column = 0; |
| 1265 | 5 | for (AlignedCodon codon : alignedCodons.keySet()) |
| 1266 | { | |
| 1267 | 1914 | final Map<SequenceI, AlignedCodon> columnResidues = alignedCodons |
| 1268 | .get(codon); | |
| 1269 | 1914 | for (Entry<SequenceI, AlignedCodon> entry : columnResidues.entrySet()) |
| 1270 | { | |
| 1271 | 10682 | char residue = entry.getValue().product.charAt(0); |
| 1272 | 10682 | peptides.get(entry.getKey())[column] = residue; |
| 1273 | } | |
| 1274 | 1914 | column++; |
| 1275 | } | |
| 1276 | ||
| 1277 | /* | |
| 1278 | * and finally set the constructed sequences | |
| 1279 | */ | |
| 1280 | 5 | for (Entry<SequenceI, char[]> entry : peptides.entrySet()) |
| 1281 | { | |
| 1282 | 30 | entry.getKey().setSequence(new String(entry.getValue())); |
| 1283 | } | |
| 1284 | ||
| 1285 | 5 | return 0; |
| 1286 | } | |
| 1287 | ||
| 1288 | /** | |
| 1289 | * Populate the map of aligned codons by traversing the given sequence | |
| 1290 | * mapping, locating the aligned positions of mapped codons, and adding those | |
| 1291 | * positions and their translation products to the map. | |
| 1292 | * | |
| 1293 | * @param dna | |
| 1294 | * the aligned sequence we are mapping from | |
| 1295 | * @param protein | |
| 1296 | * the sequence to be aligned to the codons | |
| 1297 | * @param gapChar | |
| 1298 | * the gap character in the dna sequence | |
| 1299 | * @param seqMap | |
| 1300 | * a mapping to a sequence translation | |
| 1301 | * @param alignedCodons | |
| 1302 | * the map we are building up | |
| 1303 | */ | |
| 1304 | 32 | static void addCodonPositions(SequenceI dna, SequenceI protein, |
| 1305 | char gapChar, Mapping seqMap, | |
| 1306 | Map<AlignedCodon, Map<SequenceI, AlignedCodon>> alignedCodons) | |
| 1307 | { | |
| 1308 | 32 | Iterator<AlignedCodon> codons = seqMap.getCodonIterator(dna, gapChar); |
| 1309 | ||
| 1310 | /* | |
| 1311 | * add codon positions, and their peptide translations, to the alignment | |
| 1312 | * map, while remembering the first codon mapped | |
| 1313 | */ | |
| 1314 | 10716 | while (codons.hasNext()) |
| 1315 | { | |
| 1316 | 10684 | try |
| 1317 | { | |
| 1318 | 10684 | AlignedCodon codon = codons.next(); |
| 1319 | 10684 | addCodonToMap(alignedCodons, codon, protein); |
| 1320 | } catch (IncompleteCodonException e) | |
| 1321 | { | |
| 1322 | // possible incomplete trailing codon - ignore | |
| 1323 | } catch (NoSuchElementException e) | |
| 1324 | { | |
| 1325 | // possibly peptide lacking STOP | |
| 1326 | } | |
| 1327 | } | |
| 1328 | } | |
| 1329 | ||
| 1330 | /** | |
| 1331 | * Helper method to add a codon-to-peptide entry to the aligned codons map | |
| 1332 | * | |
| 1333 | * @param alignedCodons | |
| 1334 | * @param codon | |
| 1335 | * @param protein | |
| 1336 | */ | |
| 1337 | 10690 | protected static void addCodonToMap( |
| 1338 | Map<AlignedCodon, Map<SequenceI, AlignedCodon>> alignedCodons, | |
| 1339 | AlignedCodon codon, SequenceI protein) | |
| 1340 | { | |
| 1341 | 10690 | Map<SequenceI, AlignedCodon> seqProduct = alignedCodons.get(codon); |
| 1342 | 10690 | if (seqProduct == null) |
| 1343 | { | |
| 1344 | 1914 | seqProduct = new HashMap<>(); |
| 1345 | 1914 | alignedCodons.put(codon, seqProduct); |
| 1346 | } | |
| 1347 | 10690 | seqProduct.put(protein, codon); |
| 1348 | } | |
| 1349 | ||
| 1350 | /** | |
| 1351 | * Returns true if a cDNA/Protein mapping either exists, or could be made, | |
| 1352 | * between at least one pair of sequences in the two alignments. Currently, | |
| 1353 | * the logic is: | |
| 1354 | * <ul> | |
| 1355 | * <li>One alignment must be nucleotide, and the other protein</li> | |
| 1356 | * <li>At least one pair of sequences must be already mapped, or mappable</li> | |
| 1357 | * <li>Mappable means the nucleotide translation matches the protein | |
| 1358 | * sequence</li> | |
| 1359 | * <li>The translation may ignore start and stop codons if present in the | |
| 1360 | * nucleotide</li> | |
| 1361 | * </ul> | |
| 1362 | * | |
| 1363 | * @param al1 | |
| 1364 | * @param al2 | |
| 1365 | * @return | |
| 1366 | */ | |
| 1367 | 28 | public static boolean isMappable(AlignmentI al1, AlignmentI al2) |
| 1368 | { | |
| 1369 | 28 | if (al1 == null || al2 == null) |
| 1370 | { | |
| 1371 | 3 | return false; |
| 1372 | } | |
| 1373 | ||
| 1374 | /* | |
| 1375 | * Require one nucleotide and one protein | |
| 1376 | */ | |
| 1377 | 25 | if (al1.isNucleotide() == al2.isNucleotide()) |
| 1378 | { | |
| 1379 | 14 | return false; |
| 1380 | } | |
| 1381 | 11 | AlignmentI dna = al1.isNucleotide() ? al1 : al2; |
| 1382 | 11 | AlignmentI protein = dna == al1 ? al2 : al1; |
| 1383 | 11 | List<AlignedCodonFrame> mappings = protein.getCodonFrames(); |
| 1384 | 11 | for (SequenceI dnaSeq : dna.getSequences()) |
| 1385 | { | |
| 1386 | 12 | for (SequenceI proteinSeq : protein.getSequences()) |
| 1387 | { | |
| 1388 | 12 | if (isMappable(dnaSeq, proteinSeq, mappings)) |
| 1389 | { | |
| 1390 | 2 | return true; |
| 1391 | } | |
| 1392 | } | |
| 1393 | } | |
| 1394 | 9 | return false; |
| 1395 | } | |
| 1396 | ||
| 1397 | /** | |
| 1398 | * Returns true if the dna sequence is mapped, or could be mapped, to the | |
| 1399 | * protein sequence. | |
| 1400 | * | |
| 1401 | * @param dnaSeq | |
| 1402 | * @param proteinSeq | |
| 1403 | * @param mappings | |
| 1404 | * @return | |
| 1405 | */ | |
| 1406 | 12 | protected static boolean isMappable(SequenceI dnaSeq, |
| 1407 | SequenceI proteinSeq, List<AlignedCodonFrame> mappings) | |
| 1408 | { | |
| 1409 | 12 | if (dnaSeq == null || proteinSeq == null) |
| 1410 | { | |
| 1411 | 0 | return false; |
| 1412 | } | |
| 1413 | ||
| 1414 | 12 | SequenceI dnaDs = dnaSeq.getDatasetSequence() == null ? dnaSeq |
| 1415 | : dnaSeq.getDatasetSequence(); | |
| 1416 | 12 | SequenceI proteinDs = proteinSeq.getDatasetSequence() == null |
| 1417 | ? proteinSeq | |
| 1418 | : proteinSeq.getDatasetSequence(); | |
| 1419 | ||
| 1420 | 12 | for (AlignedCodonFrame mapping : mappings) |
| 1421 | { | |
| 1422 | 0 | if (proteinDs == mapping.getAaForDnaSeq(dnaDs)) |
| 1423 | { | |
| 1424 | /* | |
| 1425 | * already mapped | |
| 1426 | */ | |
| 1427 | 0 | return true; |
| 1428 | } | |
| 1429 | } | |
| 1430 | ||
| 1431 | /* | |
| 1432 | * Just try to make a mapping (it is not yet stored), test whether | |
| 1433 | * successful. | |
| 1434 | */ | |
| 1435 | 12 | return mapCdnaToProtein(proteinDs, dnaDs) != null; |
| 1436 | } | |
| 1437 | ||
| 1438 | /** | |
| 1439 | * Finds any reference annotations associated with the sequences in | |
| 1440 | * sequenceScope, that are not already added to the alignment, and adds them | |
| 1441 | * to the 'candidates' map. Also populates a lookup table of annotation | |
| 1442 | * labels, keyed by calcId, for use in constructing tooltips or the like. | |
| 1443 | * | |
| 1444 | * @param sequenceScope | |
| 1445 | * the sequences to scan for reference annotations | |
| 1446 | * @param labelForCalcId | |
| 1447 | * (optional) map to populate with label for calcId | |
| 1448 | * @param candidates | |
| 1449 | * map to populate with annotations for sequence | |
| 1450 | * @param al | |
| 1451 | * the alignment to check for presence of annotations | |
| 1452 | */ | |
| 1453 | 86 | public static void findAddableReferenceAnnotations( |
| 1454 | List<SequenceI> sequenceScope, Map<String, String> labelForCalcId, | |
| 1455 | final Map<SequenceI, List<AlignmentAnnotation>> candidates, | |
| 1456 | AlignmentI al) | |
| 1457 | { | |
| 1458 | 86 | if (sequenceScope == null) |
| 1459 | { | |
| 1460 | 1 | return; |
| 1461 | } | |
| 1462 | ||
| 1463 | /* | |
| 1464 | * For each sequence in scope, make a list of any annotations on the | |
| 1465 | * underlying dataset sequence which are not already on the alignment. | |
| 1466 | * | |
| 1467 | * Add to a map of { alignmentSequence, <List of annotations to add> } | |
| 1468 | */ | |
| 1469 | 85 | for (SequenceI seq : sequenceScope) |
| 1470 | { | |
| 1471 | 82 | SequenceI dataset = seq.getDatasetSequence(); |
| 1472 | 82 | if (dataset == null) |
| 1473 | { | |
| 1474 | 0 | continue; |
| 1475 | } | |
| 1476 | 82 | AlignmentAnnotation[] datasetAnnotations = dataset.getAnnotation(); |
| 1477 | 82 | if (datasetAnnotations == null) |
| 1478 | { | |
| 1479 | 35 | continue; |
| 1480 | } | |
| 1481 | 47 | final List<AlignmentAnnotation> result = new ArrayList<>(); |
| 1482 | 47 | for (AlignmentAnnotation dsann : datasetAnnotations) |
| 1483 | { | |
| 1484 | /* | |
| 1485 | * Find matching annotations on the alignment. If none is found, then | |
| 1486 | * add this annotation to the list of 'addable' annotations for this | |
| 1487 | * sequence. | |
| 1488 | */ | |
| 1489 | 155 | final Iterable<AlignmentAnnotation> matchedAlignmentAnnotations = al |
| 1490 | .findAnnotations(seq, dsann.getCalcId(), dsann.label); | |
| 1491 | 155 | boolean found = false; |
| 1492 | 155 | if (matchedAlignmentAnnotations != null) |
| 1493 | { | |
| 1494 | 152 | for (AlignmentAnnotation matched : matchedAlignmentAnnotations) |
| 1495 | { | |
| 1496 | 135 | if (dsann.description.equals(matched.description)) |
| 1497 | { | |
| 1498 | 54 | found = true; |
| 1499 | 54 | break; |
| 1500 | } | |
| 1501 | } | |
| 1502 | } | |
| 1503 | 155 | if (!found) |
| 1504 | { | |
| 1505 | 101 | result.add(dsann); |
| 1506 | 101 | if (labelForCalcId != null) |
| 1507 | { | |
| 1508 | 10 | labelForCalcId.put(dsann.getCalcId(), dsann.label); |
| 1509 | } | |
| 1510 | } | |
| 1511 | } | |
| 1512 | /* | |
| 1513 | * Save any addable annotations for this sequence | |
| 1514 | */ | |
| 1515 | 47 | if (!result.isEmpty()) |
| 1516 | { | |
| 1517 | 43 | candidates.put(seq, result); |
| 1518 | } | |
| 1519 | } | |
| 1520 | } | |
| 1521 | ||
| 1522 | /** | |
| 1523 | * Adds annotations to the top of the alignment annotations, in the same order | |
| 1524 | * as their related sequences. If you already have an annotation and want to | |
| 1525 | * add it to a sequence in an alignment use {@code addReferenceAnnotationTo} | |
| 1526 | * | |
| 1527 | * @param annotations | |
| 1528 | * the annotations to add | |
| 1529 | * @param alignment | |
| 1530 | * the alignment to add them to | |
| 1531 | * @param selectionGroup | |
| 1532 | * current selection group - may be null, if provided then any added | |
| 1533 | * annotation will be trimmed to just those columns in the selection | |
| 1534 | * group | |
| 1535 | */ | |
| 1536 | 40 | public static void addReferenceAnnotations( |
| 1537 | Map<SequenceI, List<AlignmentAnnotation>> annotations, | |
| 1538 | final AlignmentI alignment, final SequenceGroup selectionGroup) | |
| 1539 | { | |
| 1540 | 40 | for (SequenceI seq : annotations.keySet()) |
| 1541 | { | |
| 1542 | 39 | for (AlignmentAnnotation ann : annotations.get(seq)) |
| 1543 | { | |
| 1544 | 95 | addReferenceAnnotationTo(alignment, seq, ann, selectionGroup); |
| 1545 | } | |
| 1546 | } | |
| 1547 | } | |
| 1548 | ||
| 1549 | 0 | public static boolean isSSAnnotationPresent( |
| 1550 | Map<SequenceI, List<AlignmentAnnotation>> annotations) | |
| 1551 | { | |
| 1552 | ||
| 1553 | 0 | for (SequenceI seq : annotations.keySet()) |
| 1554 | { | |
| 1555 | 0 | if (isSecondaryStructurePresent( |
| 1556 | annotations.get(seq).toArray(new AlignmentAnnotation[0]))) | |
| 1557 | { | |
| 1558 | 0 | return true; |
| 1559 | } | |
| 1560 | } | |
| 1561 | 0 | return false; |
| 1562 | } | |
| 1563 | ||
| 1564 | /** | |
| 1565 | * Make a copy of a reference annotation {@code ann} and add it to an | |
| 1566 | * alignment sequence {@code seq} in {@code alignment}, optionally limited to | |
| 1567 | * the extent of {@code selectionGroup} | |
| 1568 | * | |
| 1569 | * @param alignment | |
| 1570 | * @param seq | |
| 1571 | * @param ann | |
| 1572 | * @param selectionGroup | |
| 1573 | * current selection group - may be null, if provided then any added | |
| 1574 | * annotation will be trimmed to just those columns in the selection | |
| 1575 | * group | |
| 1576 | * @return annotation added to {@code seq and {@code alignment} | |
| 1577 | */ | |
| 1578 | 99 | public static AlignmentAnnotation addReferenceAnnotationTo( |
| 1579 | final AlignmentI alignment, final SequenceI seq, | |
| 1580 | final AlignmentAnnotation ann, final SequenceGroup selectionGroup) | |
| 1581 | { | |
| 1582 | 99 | AlignmentAnnotation copyAnn = new AlignmentAnnotation(ann); |
| 1583 | 99 | int startRes = 0; |
| 1584 | 99 | int endRes = ann.annotations.length; |
| 1585 | 99 | if (selectionGroup != null) |
| 1586 | { | |
| 1587 | 2 | startRes = -1 + Math.min(seq.getEnd(), Math.max(seq.getStart(), |
| 1588 | seq.findPosition(selectionGroup.getStartRes()))); | |
| 1589 | 2 | endRes = -1 + Math.min(seq.getEnd(), |
| 1590 | seq.findPosition(selectionGroup.getEndRes())); | |
| 1591 | ||
| 1592 | } | |
| 1593 | 99 | copyAnn.restrict(startRes, endRes + 0); |
| 1594 | ||
| 1595 | /* | |
| 1596 | * Add to the sequence (sets copyAnn.datasetSequence), unless the | |
| 1597 | * original annotation is already on the sequence. | |
| 1598 | */ | |
| 1599 | 99 | if (!seq.hasAnnotation(ann)) |
| 1600 | { | |
| 1601 | 99 | ContactMatrixI cm = seq.getDatasetSequence().getContactMatrixFor(ann); |
| 1602 | 99 | if (cm != null) |
| 1603 | { | |
| 1604 | 25 | seq.addContactListFor(copyAnn, cm); |
| 1605 | } | |
| 1606 | 99 | seq.addAlignmentAnnotation(copyAnn); |
| 1607 | } | |
| 1608 | // adjust for gaps | |
| 1609 | 99 | copyAnn.adjustForAlignment(); |
| 1610 | // add to the alignment and set visible | |
| 1611 | 99 | alignment.addAnnotation(copyAnn); |
| 1612 | 99 | copyAnn.visible = true; |
| 1613 | ||
| 1614 | 99 | return copyAnn; |
| 1615 | } | |
| 1616 | ||
| 1617 | /** | |
| 1618 | * Set visibility of alignment annotations of specified types (labels), for | |
| 1619 | * specified sequences. This supports controls like "Show all secondary | |
| 1620 | * structure", "Hide all Temp factor", etc. | |
| 1621 | * | |
| 1622 | * @al the alignment to scan for annotations | |
| 1623 | * @param types | |
| 1624 | * the types (labels) of annotations to be updated | |
| 1625 | * @param forSequences | |
| 1626 | * if not null, only annotations linked to one of these sequences are | |
| 1627 | * in scope for update; if null, acts on all sequence annotations | |
| 1628 | * @param anyType | |
| 1629 | * if this flag is true, 'types' is ignored (label not checked) | |
| 1630 | * @param doShow | |
| 1631 | * if true, set visibility on, else set off | |
| 1632 | */ | |
| 1633 | 5 | public static void showOrHideSequenceAnnotations(AlignmentI al, |
| 1634 | Collection<String> types, List<SequenceI> forSequences, | |
| 1635 | boolean anyType, boolean doShow) | |
| 1636 | { | |
| 1637 | 5 | AlignmentAnnotation[] anns = al.getAlignmentAnnotation(); |
| 1638 | 5 | if (anns != null) |
| 1639 | { | |
| 1640 | 5 | for (AlignmentAnnotation aa : anns) |
| 1641 | { | |
| 1642 | 30 | if (anyType || types.contains(aa.label)) |
| 1643 | { | |
| 1644 | 21 | if ((aa.sequenceRef != null) && (forSequences == null |
| 1645 | || forSequences.contains(aa.sequenceRef))) | |
| 1646 | { | |
| 1647 | 11 | aa.visible = doShow; |
| 1648 | } | |
| 1649 | } | |
| 1650 | } | |
| 1651 | } | |
| 1652 | } | |
| 1653 | ||
| 1654 | /** | |
| 1655 | * Shows or hides auto calculated annotations for a sequence group. | |
| 1656 | * | |
| 1657 | * @param al | |
| 1658 | * The alignment object with the annotations. | |
| 1659 | * @param type | |
| 1660 | * The type of annotation to show or hide. | |
| 1661 | * @param selectedGroup | |
| 1662 | * The sequence group for which the annotations should be shown or | |
| 1663 | * hidden. | |
| 1664 | * @param anyType | |
| 1665 | * If true, all types of annotations will be shown/hidden. | |
| 1666 | * @param doShow | |
| 1667 | * If true, the annotations will be shown; if false, annotations will | |
| 1668 | * be hidden. | |
| 1669 | */ | |
| 1670 | 0 | public static void showOrHideAutoCalculatedAnnotationsForGroup( |
| 1671 | AlignmentI al, String type, SequenceGroup selectedGroup, | |
| 1672 | boolean anyType, boolean doShow) | |
| 1673 | { | |
| 1674 | // Get all alignment annotations | |
| 1675 | 0 | AlignmentAnnotation[] anns = al.getAlignmentAnnotation(); |
| 1676 | ||
| 1677 | 0 | if (anns != null) |
| 1678 | { | |
| 1679 | 0 | for (AlignmentAnnotation aa : anns) |
| 1680 | { | |
| 1681 | // Check if anyType is true or if the annotation's label contains the | |
| 1682 | // specified type (currently for secondary structure consensus) | |
| 1683 | 0 | if ((anyType && aa.label |
| 1684 | .startsWith(Constants.SECONDARY_STRUCTURE_CONSENSUS_LABEL)) | |
| 1685 | || aa.label.startsWith(type)) | |
| 1686 | { | |
| 1687 | // If the annotation's group reference is not null and matches the | |
| 1688 | // selected group, update its visibility. | |
| 1689 | 0 | if (aa.groupRef != null && selectedGroup == aa.groupRef) |
| 1690 | { | |
| 1691 | 0 | aa.visible = doShow; |
| 1692 | } | |
| 1693 | } | |
| 1694 | } | |
| 1695 | } | |
| 1696 | } | |
| 1697 | ||
| 1698 | 0 | public static AlignmentAnnotation getFirstSequenceAnnotationOfType( |
| 1699 | AlignmentI al, int graphType) | |
| 1700 | { | |
| 1701 | 0 | AlignmentAnnotation[] anns = al.getAlignmentAnnotation(); |
| 1702 | 0 | if (anns != null) |
| 1703 | { | |
| 1704 | 0 | for (AlignmentAnnotation aa : anns) |
| 1705 | { | |
| 1706 | 0 | if (aa.sequenceRef != null && aa.graph == graphType) |
| 1707 | 0 | return aa; |
| 1708 | } | |
| 1709 | } | |
| 1710 | 0 | return null; |
| 1711 | } | |
| 1712 | ||
| 1713 | /** | |
| 1714 | * Returns true if either sequence has a cross-reference to the other | |
| 1715 | * | |
| 1716 | * @param seq1 | |
| 1717 | * @param seq2 | |
| 1718 | * @return | |
| 1719 | */ | |
| 1720 | 52 | public static boolean haveCrossRef(SequenceI seq1, SequenceI seq2) |
| 1721 | { | |
| 1722 | // Note: moved here from class CrossRef as the latter class has dependencies | |
| 1723 | // not availability to the applet's classpath | |
| 1724 | 52 | return hasCrossRef(seq1, seq2) || hasCrossRef(seq2, seq1); |
| 1725 | } | |
| 1726 | ||
| 1727 | /** | |
| 1728 | * Returns true if seq1 has a cross-reference to seq2. Currently this assumes | |
| 1729 | * that sequence name is structured as Source|AccessionId. | |
| 1730 | * | |
| 1731 | * @param seq1 | |
| 1732 | * @param seq2 | |
| 1733 | * @return | |
| 1734 | */ | |
| 1735 | 108 | public static boolean hasCrossRef(SequenceI seq1, SequenceI seq2) |
| 1736 | { | |
| 1737 | 108 | if (seq1 == null || seq2 == null) |
| 1738 | { | |
| 1739 | 8 | return false; |
| 1740 | } | |
| 1741 | 100 | String name = seq2.getName(); |
| 1742 | 100 | final List<DBRefEntry> xrefs = seq1.getDBRefs(); |
| 1743 | 100 | if (xrefs != null) |
| 1744 | { | |
| 1745 | 34 | for (int ix = 0, nx = xrefs.size(); ix < nx; ix++) |
| 1746 | { | |
| 1747 | 24 | DBRefEntry xref = xrefs.get(ix); |
| 1748 | 24 | String xrefName = xref.getSource() + "|" + xref.getAccessionId(); |
| 1749 | // case-insensitive test, consistent with DBRefEntry.equalRef() | |
| 1750 | 24 | if (xrefName.equalsIgnoreCase(name)) |
| 1751 | { | |
| 1752 | 12 | return true; |
| 1753 | } | |
| 1754 | } | |
| 1755 | } | |
| 1756 | 88 | return false; |
| 1757 | } | |
| 1758 | ||
| 1759 | /** | |
| 1760 | * Constructs an alignment consisting of the mapped (CDS) regions in the given | |
| 1761 | * nucleotide sequences, and updates mappings to match. The CDS sequences are | |
| 1762 | * added to the original alignment's dataset, which is shared by the new | |
| 1763 | * alignment. Mappings from nucleotide to CDS, and from CDS to protein, are | |
| 1764 | * added to the alignment dataset. | |
| 1765 | * | |
| 1766 | * @param dna | |
| 1767 | * aligned nucleotide (dna or cds) sequences | |
| 1768 | * @param dataset | |
| 1769 | * the alignment dataset the sequences belong to | |
| 1770 | * @param products | |
| 1771 | * (optional) to restrict results to CDS that map to specified | |
| 1772 | * protein products | |
| 1773 | * @return an alignment whose sequences are the cds-only parts of the dna | |
| 1774 | * sequences (or null if no mappings are found) | |
| 1775 | */ | |
| 1776 | 6 | public static AlignmentI makeCdsAlignment(SequenceI[] dna, |
| 1777 | AlignmentI dataset, SequenceI[] products) | |
| 1778 | { | |
| 1779 | 6 | if (dataset == null || dataset.getDataset() != null) |
| 1780 | { | |
| 1781 | 0 | throw new IllegalArgumentException( |
| 1782 | "IMPLEMENTATION ERROR: dataset.getDataset() must be null!"); | |
| 1783 | } | |
| 1784 | 6 | List<SequenceI> foundSeqs = new ArrayList<>(); |
| 1785 | 6 | List<SequenceI> cdsSeqs = new ArrayList<>(); |
| 1786 | 6 | List<AlignedCodonFrame> mappings = dataset.getCodonFrames(); |
| 1787 | 6 | HashSet<SequenceI> productSeqs = null; |
| 1788 | 6 | if (products != null) |
| 1789 | { | |
| 1790 | 3 | productSeqs = new HashSet<>(); |
| 1791 | 3 | for (SequenceI seq : products) |
| 1792 | { | |
| 1793 | 24 | productSeqs.add(seq.getDatasetSequence() == null ? seq |
| 1794 | : seq.getDatasetSequence()); | |
| 1795 | } | |
| 1796 | } | |
| 1797 | ||
| 1798 | /* | |
| 1799 | * Construct CDS sequences from mappings on the alignment dataset. | |
| 1800 | * The logic is: | |
| 1801 | * - find the protein product(s) mapped to from each dna sequence | |
| 1802 | * - if the mapping covers the whole dna sequence (give or take start/stop | |
| 1803 | * codon), take the dna as the CDS sequence | |
| 1804 | * - else search dataset mappings for a suitable dna sequence, i.e. one | |
| 1805 | * whose whole sequence is mapped to the protein | |
| 1806 | * - if no sequence found, construct one from the dna sequence and mapping | |
| 1807 | * (and add it to dataset so it is found if this is repeated) | |
| 1808 | */ | |
| 1809 | 6 | for (SequenceI dnaSeq : dna) |
| 1810 | { | |
| 1811 | 52 | SequenceI dnaDss = dnaSeq.getDatasetSequence() == null ? dnaSeq |
| 1812 | : dnaSeq.getDatasetSequence(); | |
| 1813 | ||
| 1814 | 52 | List<AlignedCodonFrame> seqMappings = MappingUtils |
| 1815 | .findMappingsForSequence(dnaSeq, mappings); | |
| 1816 | 52 | for (AlignedCodonFrame mapping : seqMappings) |
| 1817 | { | |
| 1818 | 42 | List<Mapping> mappingsFromSequence = mapping |
| 1819 | .getMappingsFromSequence(dnaSeq); | |
| 1820 | ||
| 1821 | 42 | for (Mapping aMapping : mappingsFromSequence) |
| 1822 | { | |
| 1823 | 44 | MapList mapList = aMapping.getMap(); |
| 1824 | 44 | if (mapList.getFromRatio() == 1) |
| 1825 | { | |
| 1826 | /* | |
| 1827 | * not a dna-to-protein mapping (likely dna-to-cds) | |
| 1828 | */ | |
| 1829 | 11 | continue; |
| 1830 | } | |
| 1831 | ||
| 1832 | /* | |
| 1833 | * skip if mapping is not to one of the target set of proteins | |
| 1834 | */ | |
| 1835 | 33 | SequenceI proteinProduct = aMapping.getTo(); |
| 1836 | 33 | if (productSeqs != null && !productSeqs.contains(proteinProduct)) |
| 1837 | { | |
| 1838 | 2 | continue; |
| 1839 | } | |
| 1840 | ||
| 1841 | /* | |
| 1842 | * try to locate the CDS from the dataset mappings; | |
| 1843 | * guard against duplicate results (for the case that protein has | |
| 1844 | * dbrefs to both dna and cds sequences) | |
| 1845 | */ | |
| 1846 | 31 | SequenceI cdsSeq = findCdsForProtein(mappings, dnaSeq, |
| 1847 | seqMappings, aMapping); | |
| 1848 | 31 | if (cdsSeq != null) |
| 1849 | { | |
| 1850 | 11 | if (!foundSeqs.contains(cdsSeq)) |
| 1851 | { | |
| 1852 | 11 | foundSeqs.add(cdsSeq); |
| 1853 | 11 | SequenceI derivedSequence = cdsSeq.deriveSequence(); |
| 1854 | 11 | cdsSeqs.add(derivedSequence); |
| 1855 | 11 | if (!dataset.getSequences().contains(cdsSeq)) |
| 1856 | { | |
| 1857 | 0 | dataset.addSequence(cdsSeq); |
| 1858 | } | |
| 1859 | } | |
| 1860 | 11 | continue; |
| 1861 | } | |
| 1862 | ||
| 1863 | /* | |
| 1864 | * didn't find mapped CDS sequence - construct it and add | |
| 1865 | * its dataset sequence to the dataset | |
| 1866 | */ | |
| 1867 | 20 | cdsSeq = makeCdsSequence(dnaSeq.getDatasetSequence(), aMapping, |
| 1868 | dataset).deriveSequence(); | |
| 1869 | // cdsSeq has a name constructed as CDS|<dbref> | |
| 1870 | // <dbref> will be either the accession for the coding sequence, | |
| 1871 | // marked in the /via/ dbref to the protein product accession | |
| 1872 | // or it will be the original nucleotide accession. | |
| 1873 | 20 | SequenceI cdsSeqDss = cdsSeq.getDatasetSequence(); |
| 1874 | ||
| 1875 | 20 | cdsSeqs.add(cdsSeq); |
| 1876 | ||
| 1877 | /* | |
| 1878 | * build the mapping from CDS to protein | |
| 1879 | */ | |
| 1880 | 20 | List<int[]> cdsRange = Collections |
| 1881 | .singletonList(new int[] | |
| 1882 | { cdsSeq.getStart(), | |
| 1883 | cdsSeq.getLength() + cdsSeq.getStart() - 1 }); | |
| 1884 | 20 | MapList cdsToProteinMap = new MapList(cdsRange, |
| 1885 | mapList.getToRanges(), mapList.getFromRatio(), | |
| 1886 | mapList.getToRatio()); | |
| 1887 | ||
| 1888 | 20 | if (!dataset.getSequences().contains(cdsSeqDss)) |
| 1889 | { | |
| 1890 | /* | |
| 1891 | * if this sequence is a newly created one, add it to the dataset | |
| 1892 | * and made a CDS to protein mapping (if sequence already exists, | |
| 1893 | * CDS-to-protein mapping _is_ the transcript-to-protein mapping) | |
| 1894 | */ | |
| 1895 | 20 | dataset.addSequence(cdsSeqDss); |
| 1896 | 20 | AlignedCodonFrame cdsToProteinMapping = new AlignedCodonFrame(); |
| 1897 | 20 | cdsToProteinMapping.addMap(cdsSeqDss, proteinProduct, |
| 1898 | cdsToProteinMap); | |
| 1899 | ||
| 1900 | /* | |
| 1901 | * guard against duplicating the mapping if repeating this action | |
| 1902 | */ | |
| 1903 | 20 | if (!mappings.contains(cdsToProteinMapping)) |
| 1904 | { | |
| 1905 | 20 | mappings.add(cdsToProteinMapping); |
| 1906 | } | |
| 1907 | } | |
| 1908 | ||
| 1909 | 20 | propagateDBRefsToCDS(cdsSeqDss, dnaSeq.getDatasetSequence(), |
| 1910 | proteinProduct, aMapping); | |
| 1911 | /* | |
| 1912 | * add another mapping from original 'from' range to CDS | |
| 1913 | */ | |
| 1914 | 20 | AlignedCodonFrame dnaToCdsMapping = new AlignedCodonFrame(); |
| 1915 | 20 | final MapList dnaToCdsMap = new MapList(mapList.getFromRanges(), |
| 1916 | cdsRange, 1, 1); | |
| 1917 | 20 | dnaToCdsMapping.addMap(dnaSeq.getDatasetSequence(), cdsSeqDss, |
| 1918 | dnaToCdsMap); | |
| 1919 | 20 | if (!mappings.contains(dnaToCdsMapping)) |
| 1920 | { | |
| 1921 | 20 | mappings.add(dnaToCdsMapping); |
| 1922 | } | |
| 1923 | ||
| 1924 | /* | |
| 1925 | * transfer dna chromosomal loci (if known) to the CDS | |
| 1926 | * sequence (via the mapping) | |
| 1927 | */ | |
| 1928 | 20 | final MapList cdsToDnaMap = dnaToCdsMap.getInverse(); |
| 1929 | 20 | transferGeneLoci(dnaSeq, cdsToDnaMap, cdsSeq); |
| 1930 | ||
| 1931 | /* | |
| 1932 | * add DBRef with mapping from protein to CDS | |
| 1933 | * (this enables Get Cross-References from protein alignment) | |
| 1934 | * This is tricky because we can't have two DBRefs with the | |
| 1935 | * same source and accession, so need a different accession for | |
| 1936 | * the CDS from the dna sequence | |
| 1937 | */ | |
| 1938 | ||
| 1939 | // specific use case: | |
| 1940 | // Genomic contig ENSCHR:1, contains coding regions for ENSG01, | |
| 1941 | // ENSG02, ENSG03, with transcripts and products similarly named. | |
| 1942 | // cannot add distinct dbrefs mapping location on ENSCHR:1 to ENSG01 | |
| 1943 | ||
| 1944 | // JBPNote: ?? can't actually create an example that demonstrates we | |
| 1945 | // need to | |
| 1946 | // synthesize an xref. | |
| 1947 | ||
| 1948 | 20 | List<DBRefEntry> primrefs = dnaDss.getPrimaryDBRefs(); |
| 1949 | 33 | for (int ip = 0, np = primrefs.size(); ip < np; ip++) |
| 1950 | { | |
| 1951 | 13 | DBRefEntry primRef = primrefs.get(ip); |
| 1952 | /* | |
| 1953 | * create a cross-reference from CDS to the source sequence's | |
| 1954 | * primary reference and vice versa | |
| 1955 | */ | |
| 1956 | 13 | String source = primRef.getSource(); |
| 1957 | 13 | String version = primRef.getVersion(); |
| 1958 | 13 | DBRefEntry cdsCrossRef = new DBRefEntry(source, |
| 1959 | source + ":" + version, primRef.getAccessionId()); | |
| 1960 | 13 | cdsCrossRef |
| 1961 | .setMap(new Mapping(dnaDss, new MapList(cdsToDnaMap))); | |
| 1962 | 13 | cdsSeqDss.addDBRef(cdsCrossRef); |
| 1963 | ||
| 1964 | 13 | dnaSeq.addDBRef(new DBRefEntry(source, version, |
| 1965 | cdsSeq.getName(), new Mapping(cdsSeqDss, dnaToCdsMap))); | |
| 1966 | // problem here is that the cross-reference is synthesized - | |
| 1967 | // cdsSeq.getName() may be like 'CDS|dnaaccession' or | |
| 1968 | // 'CDS|emblcdsacc' | |
| 1969 | // assuming cds version same as dna ?!? | |
| 1970 | ||
| 1971 | 13 | DBRefEntry proteinToCdsRef = new DBRefEntry(source, version, |
| 1972 | cdsSeq.getName()); | |
| 1973 | // | |
| 1974 | 13 | proteinToCdsRef.setMap( |
| 1975 | new Mapping(cdsSeqDss, cdsToProteinMap.getInverse())); | |
| 1976 | 13 | proteinProduct.addDBRef(proteinToCdsRef); |
| 1977 | } | |
| 1978 | /* | |
| 1979 | * transfer any features on dna that overlap the CDS | |
| 1980 | */ | |
| 1981 | 20 | transferFeatures(dnaSeq, cdsSeq, dnaToCdsMap, null, |
| 1982 | SequenceOntologyI.CDS); | |
| 1983 | } | |
| 1984 | } | |
| 1985 | } | |
| 1986 | ||
| 1987 | 6 | AlignmentI cds = new Alignment( |
| 1988 | cdsSeqs.toArray(new SequenceI[cdsSeqs.size()])); | |
| 1989 | 6 | cds.setDataset(dataset); |
| 1990 | ||
| 1991 | 6 | return cds; |
| 1992 | } | |
| 1993 | ||
| 1994 | /** | |
| 1995 | * Tries to transfer gene loci (dbref to chromosome positions) from fromSeq to | |
| 1996 | * toSeq, mediated by the given mapping between the sequences | |
| 1997 | * | |
| 1998 | * @param fromSeq | |
| 1999 | * @param targetToFrom | |
| 2000 | * Map | |
| 2001 | * @param targetSeq | |
| 2002 | */ | |
| 2003 | 23 | protected static void transferGeneLoci(SequenceI fromSeq, |
| 2004 | MapList targetToFrom, SequenceI targetSeq) | |
| 2005 | { | |
| 2006 | 23 | if (targetSeq.getGeneLoci() != null) |
| 2007 | { | |
| 2008 | // already have - don't override | |
| 2009 | 1 | return; |
| 2010 | } | |
| 2011 | 22 | GeneLociI fromLoci = fromSeq.getGeneLoci(); |
| 2012 | 22 | if (fromLoci == null) |
| 2013 | { | |
| 2014 | 10 | return; |
| 2015 | } | |
| 2016 | ||
| 2017 | 12 | MapList newMap = targetToFrom.traverse(fromLoci.getMapping()); |
| 2018 | ||
| 2019 | 12 | if (newMap != null) |
| 2020 | { | |
| 2021 | 12 | targetSeq.setGeneLoci(fromLoci.getSpeciesId(), |
| 2022 | fromLoci.getAssemblyId(), fromLoci.getChromosomeId(), newMap); | |
| 2023 | } | |
| 2024 | } | |
| 2025 | ||
| 2026 | /** | |
| 2027 | * A helper method that finds a CDS sequence in the alignment dataset that is | |
| 2028 | * mapped to the given protein sequence, and either is, or has a mapping from, | |
| 2029 | * the given dna sequence. | |
| 2030 | * | |
| 2031 | * @param mappings | |
| 2032 | * set of all mappings on the dataset | |
| 2033 | * @param dnaSeq | |
| 2034 | * a dna (or cds) sequence we are searching from | |
| 2035 | * @param seqMappings | |
| 2036 | * the set of mappings involving dnaSeq | |
| 2037 | * @param aMapping | |
| 2038 | * a transcript-to-peptide mapping | |
| 2039 | * @return | |
| 2040 | */ | |
| 2041 | 38 | static SequenceI findCdsForProtein(List<AlignedCodonFrame> mappings, |
| 2042 | SequenceI dnaSeq, List<AlignedCodonFrame> seqMappings, | |
| 2043 | Mapping aMapping) | |
| 2044 | { | |
| 2045 | /* | |
| 2046 | * TODO a better dna-cds-protein mapping data representation to allow easy | |
| 2047 | * navigation; until then this clunky looping around lists of mappings | |
| 2048 | */ | |
| 2049 | 38 | SequenceI seqDss = dnaSeq.getDatasetSequence() == null ? dnaSeq |
| 2050 | : dnaSeq.getDatasetSequence(); | |
| 2051 | 38 | SequenceI proteinProduct = aMapping.getTo(); |
| 2052 | ||
| 2053 | /* | |
| 2054 | * is this mapping from the whole dna sequence (i.e. CDS)? | |
| 2055 | * allowing for possible stop codon on dna but not peptide | |
| 2056 | */ | |
| 2057 | 38 | int mappedFromLength = MappingUtils |
| 2058 | .getLength(aMapping.getMap().getFromRanges()); | |
| 2059 | 38 | int dnaLength = seqDss.getLength(); |
| 2060 | 38 | if (mappedFromLength == dnaLength |
| 2061 | || mappedFromLength == dnaLength - CODON_LENGTH) | |
| 2062 | { | |
| 2063 | /* | |
| 2064 | * if sequence has CDS features, this is a transcript with no UTR | |
| 2065 | * - do not take this as the CDS sequence! (JAL-2789) | |
| 2066 | */ | |
| 2067 | 4 | if (seqDss.getFeatures().getFeaturesByOntology(SequenceOntologyI.CDS) |
| 2068 | .isEmpty()) | |
| 2069 | { | |
| 2070 | 1 | return seqDss; |
| 2071 | } | |
| 2072 | } | |
| 2073 | ||
| 2074 | /* | |
| 2075 | * looks like we found the dna-to-protein mapping; search for the | |
| 2076 | * corresponding cds-to-protein mapping | |
| 2077 | */ | |
| 2078 | 37 | List<AlignedCodonFrame> mappingsToPeptide = MappingUtils |
| 2079 | .findMappingsForSequence(proteinProduct, mappings); | |
| 2080 | 37 | for (AlignedCodonFrame acf : mappingsToPeptide) |
| 2081 | { | |
| 2082 | 52 | for (SequenceToSequenceMapping map : acf.getMappings()) |
| 2083 | { | |
| 2084 | 276 | Mapping mapping = map.getMapping(); |
| 2085 | 276 | if (mapping != aMapping |
| 2086 | && mapping.getMap().getFromRatio() == CODON_LENGTH | |
| 2087 | && proteinProduct == mapping.getTo() | |
| 2088 | && seqDss != map.getFromSeq()) | |
| 2089 | { | |
| 2090 | 15 | mappedFromLength = MappingUtils |
| 2091 | .getLength(mapping.getMap().getFromRanges()); | |
| 2092 | 15 | if (mappedFromLength == map.getFromSeq().getLength()) |
| 2093 | { | |
| 2094 | /* | |
| 2095 | * found a 3:1 mapping to the protein product which covers | |
| 2096 | * the whole dna sequence i.e. is from CDS; finally check the CDS | |
| 2097 | * is mapped from the given dna start sequence | |
| 2098 | */ | |
| 2099 | 15 | SequenceI cdsSeq = map.getFromSeq(); |
| 2100 | // todo this test is weak if seqMappings contains multiple mappings; | |
| 2101 | // we get away with it if transcript:cds relationship is 1:1 | |
| 2102 | 15 | List<AlignedCodonFrame> dnaToCdsMaps = MappingUtils |
| 2103 | .findMappingsForSequence(cdsSeq, seqMappings); | |
| 2104 | 15 | if (!dnaToCdsMaps.isEmpty()) |
| 2105 | { | |
| 2106 | 13 | return cdsSeq; |
| 2107 | } | |
| 2108 | } | |
| 2109 | } | |
| 2110 | } | |
| 2111 | } | |
| 2112 | 24 | return null; |
| 2113 | } | |
| 2114 | ||
| 2115 | /** | |
| 2116 | * Helper method that makes a CDS sequence as defined by the mappings from the | |
| 2117 | * given sequence i.e. extracts the 'mapped from' ranges (which may be on | |
| 2118 | * forward or reverse strand). | |
| 2119 | * | |
| 2120 | * @param seq | |
| 2121 | * @param mapping | |
| 2122 | * @param dataset | |
| 2123 | * - existing dataset. We check for sequences that look like the CDS | |
| 2124 | * we are about to construct, if one exists already, then we will | |
| 2125 | * just return that one. | |
| 2126 | * @return CDS sequence (as a dataset sequence) | |
| 2127 | */ | |
| 2128 | 20 | static SequenceI makeCdsSequence(SequenceI seq, Mapping mapping, |
| 2129 | AlignmentI dataset) | |
| 2130 | { | |
| 2131 | /* | |
| 2132 | * construct CDS sequence name as "CDS|" with 'from id' held in the mapping | |
| 2133 | * if set (e.g. EMBL protein_id), else sequence name appended | |
| 2134 | */ | |
| 2135 | 20 | String mapFromId = mapping.getMappedFromId(); |
| 2136 | 20 | final String seqId = "CDS|" |
| 2137 | 20 | + (mapFromId != null ? mapFromId : seq.getName()); |
| 2138 | ||
| 2139 | 20 | SequenceI newSeq = null; |
| 2140 | ||
| 2141 | /* | |
| 2142 | * construct CDS sequence by splicing mapped from ranges | |
| 2143 | */ | |
| 2144 | 20 | char[] seqChars = seq.getSequence(); |
| 2145 | 20 | List<int[]> fromRanges = mapping.getMap().getFromRanges(); |
| 2146 | 20 | int cdsWidth = MappingUtils.getLength(fromRanges); |
| 2147 | 20 | char[] newSeqChars = new char[cdsWidth]; |
| 2148 | ||
| 2149 | 20 | int newPos = 0; |
| 2150 | 20 | for (int[] range : fromRanges) |
| 2151 | { | |
| 2152 | 32 | if (range[0] <= range[1]) |
| 2153 | { | |
| 2154 | // forward strand mapping - just copy the range | |
| 2155 | 32 | int length = range[1] - range[0] + 1; |
| 2156 | 32 | System.arraycopy(seqChars, range[0] - 1, newSeqChars, newPos, |
| 2157 | length); | |
| 2158 | 32 | newPos += length; |
| 2159 | } | |
| 2160 | else | |
| 2161 | { | |
| 2162 | // reverse strand mapping - copy and complement one by one | |
| 2163 | 0 | for (int i = range[0]; i >= range[1]; i--) |
| 2164 | { | |
| 2165 | 0 | newSeqChars[newPos++] = Dna.getComplement(seqChars[i - 1]); |
| 2166 | } | |
| 2167 | } | |
| 2168 | ||
| 2169 | 32 | newSeq = new Sequence(seqId, newSeqChars, 1, newPos); |
| 2170 | } | |
| 2171 | ||
| 2172 | 20 | if (dataset != null) |
| 2173 | { | |
| 2174 | 20 | SequenceI[] matches = dataset.findSequenceMatch(newSeq.getName()); |
| 2175 | 20 | if (matches != null) |
| 2176 | { | |
| 2177 | 20 | boolean matched = false; |
| 2178 | 20 | for (SequenceI mtch : matches) |
| 2179 | { | |
| 2180 | 3 | if (mtch.getStart() != newSeq.getStart()) |
| 2181 | { | |
| 2182 | 0 | continue; |
| 2183 | } | |
| 2184 | 3 | if (mtch.getEnd() != newSeq.getEnd()) |
| 2185 | { | |
| 2186 | 0 | continue; |
| 2187 | } | |
| 2188 | 3 | if (!Arrays.equals(mtch.getSequence(), newSeq.getSequence())) |
| 2189 | { | |
| 2190 | 3 | continue; |
| 2191 | } | |
| 2192 | 0 | if (!matched) |
| 2193 | { | |
| 2194 | 0 | matched = true; |
| 2195 | 0 | newSeq = mtch; |
| 2196 | } | |
| 2197 | else | |
| 2198 | { | |
| 2199 | 0 | Console.error( |
| 2200 | "JAL-2154 regression: warning - found (and ignored) a duplicate CDS sequence:" | |
| 2201 | + mtch.toString()); | |
| 2202 | } | |
| 2203 | } | |
| 2204 | } | |
| 2205 | } | |
| 2206 | // newSeq.setDescription(mapFromId); | |
| 2207 | ||
| 2208 | 20 | return newSeq; |
| 2209 | } | |
| 2210 | ||
| 2211 | /** | |
| 2212 | * Adds any DBRefEntrys to cdsSeq from contig that have a Mapping congruent to | |
| 2213 | * the given mapping. | |
| 2214 | * | |
| 2215 | * @param cdsSeq | |
| 2216 | * @param contig | |
| 2217 | * @param proteinProduct | |
| 2218 | * @param mapping | |
| 2219 | * @return list of DBRefEntrys added | |
| 2220 | */ | |
| 2221 | 20 | protected static List<DBRefEntry> propagateDBRefsToCDS(SequenceI cdsSeq, |
| 2222 | SequenceI contig, SequenceI proteinProduct, Mapping mapping) | |
| 2223 | { | |
| 2224 | ||
| 2225 | // gather direct refs from contig congruent with mapping | |
| 2226 | 20 | List<DBRefEntry> direct = new ArrayList<>(); |
| 2227 | 20 | HashSet<String> directSources = new HashSet<>(); |
| 2228 | ||
| 2229 | 20 | List<DBRefEntry> refs = contig.getDBRefs(); |
| 2230 | 20 | if (refs != null) |
| 2231 | { | |
| 2232 | 292 | for (int ib = 0, nb = refs.size(); ib < nb; ib++) |
| 2233 | { | |
| 2234 | 279 | DBRefEntry dbr = refs.get(ib); |
| 2235 | 279 | MapList map; |
| 2236 | ? | if (dbr.hasMap() && (map = dbr.getMap().getMap()).isTripletMap()) |
| 2237 | { | |
| 2238 | // check if map is the CDS mapping | |
| 2239 | 24 | if (mapping.getMap().equals(map)) |
| 2240 | { | |
| 2241 | 24 | direct.add(dbr); |
| 2242 | 24 | directSources.add(dbr.getSource()); |
| 2243 | } | |
| 2244 | } | |
| 2245 | } | |
| 2246 | } | |
| 2247 | 20 | List<DBRefEntry> onSource = DBRefUtils.selectRefs( |
| 2248 | proteinProduct.getDBRefs(), | |
| 2249 | directSources.toArray(new String[0])); | |
| 2250 | 20 | List<DBRefEntry> propagated = new ArrayList<>(); |
| 2251 | ||
| 2252 | // and generate appropriate mappings | |
| 2253 | 44 | for (int ic = 0, nc = direct.size(); ic < nc; ic++) |
| 2254 | { | |
| 2255 | 24 | DBRefEntry cdsref = direct.get(ic); |
| 2256 | 24 | Mapping m = cdsref.getMap(); |
| 2257 | // clone maplist and mapping | |
| 2258 | 24 | MapList cdsposmap = new MapList( |
| 2259 | Arrays.asList(new int[][] | |
| 2260 | { new int[] { cdsSeq.getStart(), cdsSeq.getEnd() } }), | |
| 2261 | m.getMap().getToRanges(), 3, 1); | |
| 2262 | 24 | Mapping cdsmap = new Mapping(m.getTo(), m.getMap()); |
| 2263 | ||
| 2264 | // create dbref | |
| 2265 | 24 | DBRefEntry newref = new DBRefEntry(cdsref.getSource(), |
| 2266 | cdsref.getVersion(), cdsref.getAccessionId(), | |
| 2267 | new Mapping(cdsmap.getTo(), cdsposmap)); | |
| 2268 | ||
| 2269 | // and see if we can map to the protein product for this mapping. | |
| 2270 | // onSource is the filtered set of accessions on protein that we are | |
| 2271 | // tranferring, so we assume accession is the same. | |
| 2272 | 24 | if (cdsmap.getTo() == null && onSource != null) |
| 2273 | { | |
| 2274 | 2 | List<DBRefEntry> sourceRefs = DBRefUtils.searchRefs(onSource, |
| 2275 | cdsref.getAccessionId()); | |
| 2276 | 2 | if (sourceRefs != null) |
| 2277 | { | |
| 2278 | 2 | for (DBRefEntry srcref : sourceRefs) |
| 2279 | { | |
| 2280 | 2 | if (srcref.getSource().equalsIgnoreCase(cdsref.getSource())) |
| 2281 | { | |
| 2282 | // we have found a complementary dbref on the protein product, so | |
| 2283 | // update mapping's getTo | |
| 2284 | 2 | newref.getMap().setTo(proteinProduct); |
| 2285 | } | |
| 2286 | } | |
| 2287 | } | |
| 2288 | } | |
| 2289 | 24 | cdsSeq.addDBRef(newref); |
| 2290 | 24 | propagated.add(newref); |
| 2291 | } | |
| 2292 | 20 | return propagated; |
| 2293 | } | |
| 2294 | ||
| 2295 | /** | |
| 2296 | * Transfers co-located features on 'fromSeq' to 'toSeq', adjusting the | |
| 2297 | * feature start/end ranges, optionally omitting specified feature types. | |
| 2298 | * Returns the number of features copied. | |
| 2299 | * | |
| 2300 | * @param fromSeq | |
| 2301 | * @param toSeq | |
| 2302 | * @param mapping | |
| 2303 | * the mapping from 'fromSeq' to 'toSeq' | |
| 2304 | * @param select | |
| 2305 | * if not null, only features of this type are copied (including | |
| 2306 | * subtypes in the Sequence Ontology) | |
| 2307 | * @param omitting | |
| 2308 | */ | |
| 2309 | 23 | protected static int transferFeatures(SequenceI fromSeq, SequenceI toSeq, |
| 2310 | MapList mapping, String select, String... omitting) | |
| 2311 | { | |
| 2312 | 23 | SequenceI copyTo = toSeq; |
| 2313 | 43 | while (copyTo.getDatasetSequence() != null) |
| 2314 | { | |
| 2315 | 20 | copyTo = copyTo.getDatasetSequence(); |
| 2316 | } | |
| 2317 | 23 | if (fromSeq == copyTo || fromSeq.getDatasetSequence() == copyTo) |
| 2318 | { | |
| 2319 | 0 | return 0; // shared dataset sequence |
| 2320 | } | |
| 2321 | ||
| 2322 | /* | |
| 2323 | * get features, optionally restricted by an ontology term | |
| 2324 | */ | |
| 2325 | 23 | List<SequenceFeature> sfs = select == null |
| 2326 | ? fromSeq.getFeatures().getPositionalFeatures() | |
| 2327 | : fromSeq.getFeatures().getFeaturesByOntology(select); | |
| 2328 | ||
| 2329 | 23 | int count = 0; |
| 2330 | 23 | for (SequenceFeature sf : sfs) |
| 2331 | { | |
| 2332 | 9610 | String type = sf.getType(); |
| 2333 | 9610 | boolean omit = false; |
| 2334 | 9610 | for (String toOmit : omitting) |
| 2335 | { | |
| 2336 | 9603 | if (type.equals(toOmit)) |
| 2337 | { | |
| 2338 | 134 | omit = true; |
| 2339 | } | |
| 2340 | } | |
| 2341 | 9610 | if (omit) |
| 2342 | { | |
| 2343 | 134 | continue; |
| 2344 | } | |
| 2345 | ||
| 2346 | /* | |
| 2347 | * locate the mapped range - null if either start or end is | |
| 2348 | * not mapped (no partial overlaps are calculated) | |
| 2349 | */ | |
| 2350 | 9476 | int start = sf.getBegin(); |
| 2351 | 9476 | int end = sf.getEnd(); |
| 2352 | 9476 | int[] mappedTo = mapping.locateInTo(start, end); |
| 2353 | /* | |
| 2354 | * if whole exon range doesn't map, try interpreting it | |
| 2355 | * as 5' or 3' exon overlapping the CDS range | |
| 2356 | */ | |
| 2357 | 9476 | if (mappedTo == null) |
| 2358 | { | |
| 2359 | 4447 | mappedTo = mapping.locateInTo(end, end); |
| 2360 | 4447 | if (mappedTo != null) |
| 2361 | { | |
| 2362 | /* | |
| 2363 | * end of exon is in CDS range - 5' overlap | |
| 2364 | * to a range from the start of the peptide | |
| 2365 | */ | |
| 2366 | 0 | mappedTo[0] = 1; |
| 2367 | } | |
| 2368 | } | |
| 2369 | 9476 | if (mappedTo == null) |
| 2370 | { | |
| 2371 | 4447 | mappedTo = mapping.locateInTo(start, start); |
| 2372 | 4447 | if (mappedTo != null) |
| 2373 | { | |
| 2374 | /* | |
| 2375 | * start of exon is in CDS range - 3' overlap | |
| 2376 | * to a range up to the end of the peptide | |
| 2377 | */ | |
| 2378 | 0 | mappedTo[1] = toSeq.getLength(); |
| 2379 | } | |
| 2380 | } | |
| 2381 | 9476 | if (mappedTo != null) |
| 2382 | { | |
| 2383 | 5029 | int newBegin = Math.min(mappedTo[0], mappedTo[1]); |
| 2384 | 5029 | int newEnd = Math.max(mappedTo[0], mappedTo[1]); |
| 2385 | 5029 | SequenceFeature copy = new SequenceFeature(sf, newBegin, newEnd, |
| 2386 | sf.getFeatureGroup(), sf.getScore()); | |
| 2387 | 5029 | copyTo.addSequenceFeature(copy); |
| 2388 | 5029 | count++; |
| 2389 | } | |
| 2390 | } | |
| 2391 | 23 | return count; |
| 2392 | } | |
| 2393 | ||
| 2394 | /** | |
| 2395 | * Returns a mapping from dna to protein by inspecting sequence features of | |
| 2396 | * type "CDS" on the dna. A mapping is constructed if the total CDS feature | |
| 2397 | * length is 3 times the peptide length (optionally after dropping a trailing | |
| 2398 | * stop codon). This method does not check whether the CDS nucleotide sequence | |
| 2399 | * translates to the peptide sequence. | |
| 2400 | * | |
| 2401 | * @param dnaSeq | |
| 2402 | * @param proteinSeq | |
| 2403 | * @return | |
| 2404 | */ | |
| 2405 | 22 | public static MapList mapCdsToProtein(SequenceI dnaSeq, |
| 2406 | SequenceI proteinSeq) | |
| 2407 | { | |
| 2408 | 22 | List<int[]> ranges = findCdsPositions(dnaSeq); |
| 2409 | 22 | int mappedDnaLength = MappingUtils.getLength(ranges); |
| 2410 | ||
| 2411 | /* | |
| 2412 | * if not a whole number of codons, truncate mapping | |
| 2413 | */ | |
| 2414 | 22 | int codonRemainder = mappedDnaLength % CODON_LENGTH; |
| 2415 | 22 | if (codonRemainder > 0) |
| 2416 | { | |
| 2417 | 2 | mappedDnaLength -= codonRemainder; |
| 2418 | 2 | MappingUtils.removeEndPositions(codonRemainder, ranges); |
| 2419 | } | |
| 2420 | ||
| 2421 | 22 | int proteinLength = proteinSeq.getLength(); |
| 2422 | 22 | int proteinStart = proteinSeq.getStart(); |
| 2423 | 22 | int proteinEnd = proteinSeq.getEnd(); |
| 2424 | ||
| 2425 | /* | |
| 2426 | * incomplete start codon may mean X at start of peptide | |
| 2427 | * we ignore both for mapping purposes | |
| 2428 | */ | |
| 2429 | 22 | if (proteinSeq.getCharAt(0) == 'X') |
| 2430 | { | |
| 2431 | // todo JAL-2022 support startPhase > 0 | |
| 2432 | 1 | proteinStart++; |
| 2433 | 1 | proteinLength--; |
| 2434 | } | |
| 2435 | 22 | List<int[]> proteinRange = new ArrayList<>(); |
| 2436 | ||
| 2437 | /* | |
| 2438 | * dna length should map to protein (or protein plus stop codon) | |
| 2439 | */ | |
| 2440 | 22 | int codesForResidues = mappedDnaLength / CODON_LENGTH; |
| 2441 | 22 | if (codesForResidues == (proteinLength + 1)) |
| 2442 | { | |
| 2443 | // assuming extra codon is for STOP and not in peptide | |
| 2444 | // todo: check trailing codon is indeed a STOP codon | |
| 2445 | 2 | codesForResidues--; |
| 2446 | 2 | mappedDnaLength -= CODON_LENGTH; |
| 2447 | 2 | MappingUtils.removeEndPositions(CODON_LENGTH, ranges); |
| 2448 | } | |
| 2449 | ||
| 2450 | 22 | if (codesForResidues == proteinLength) |
| 2451 | { | |
| 2452 | 4 | proteinRange.add(new int[] { proteinStart, proteinEnd }); |
| 2453 | 4 | return new MapList(ranges, proteinRange, CODON_LENGTH, 1); |
| 2454 | } | |
| 2455 | 18 | return null; |
| 2456 | } | |
| 2457 | ||
| 2458 | /** | |
| 2459 | * Returns a list of CDS ranges found (as sequence positions base 1), i.e. of | |
| 2460 | * [start, end] positions of sequence features of type "CDS" (or a sub-type of | |
| 2461 | * CDS in the Sequence Ontology). The ranges are sorted into ascending start | |
| 2462 | * position order, so this method is only valid for linear CDS in the same | |
| 2463 | * sense as the protein product. | |
| 2464 | * | |
| 2465 | * @param dnaSeq | |
| 2466 | * @return | |
| 2467 | */ | |
| 2468 | 24 | protected static List<int[]> findCdsPositions(SequenceI dnaSeq) |
| 2469 | { | |
| 2470 | 24 | List<int[]> result = new ArrayList<>(); |
| 2471 | ||
| 2472 | 24 | List<SequenceFeature> sfs = dnaSeq.getFeatures() |
| 2473 | .getFeaturesByOntology(SequenceOntologyI.CDS); | |
| 2474 | 24 | if (sfs.isEmpty()) |
| 2475 | { | |
| 2476 | 16 | return result; |
| 2477 | } | |
| 2478 | 8 | SequenceFeatures.sortFeatures(sfs, true); |
| 2479 | ||
| 2480 | 8 | for (SequenceFeature sf : sfs) |
| 2481 | { | |
| 2482 | 16 | int phase = 0; |
| 2483 | 16 | try |
| 2484 | { | |
| 2485 | 16 | String s = sf.getPhase(); |
| 2486 | 16 | if (s != null) |
| 2487 | { | |
| 2488 | 2 | phase = Integer.parseInt(s); |
| 2489 | } | |
| 2490 | } catch (NumberFormatException e) | |
| 2491 | { | |
| 2492 | // leave as zero | |
| 2493 | } | |
| 2494 | /* | |
| 2495 | * phase > 0 on first codon means 5' incomplete - skip to the start | |
| 2496 | * of the next codon; example ENST00000496384 | |
| 2497 | */ | |
| 2498 | 16 | int begin = sf.getBegin(); |
| 2499 | 16 | int end = sf.getEnd(); |
| 2500 | 16 | if (result.isEmpty() && phase > 0) |
| 2501 | { | |
| 2502 | 2 | begin += phase; |
| 2503 | 2 | if (begin > end) |
| 2504 | { | |
| 2505 | // shouldn't happen! | |
| 2506 | 0 | System.err |
| 2507 | .println("Error: start phase extends beyond start CDS in " | |
| 2508 | + dnaSeq.getName()); | |
| 2509 | } | |
| 2510 | } | |
| 2511 | 16 | result.add(new int[] { begin, end }); |
| 2512 | } | |
| 2513 | ||
| 2514 | /* | |
| 2515 | * Finally sort ranges by start position. This avoids a dependency on | |
| 2516 | * keeping features in order on the sequence (if they are in order anyway, | |
| 2517 | * the sort will have almost no work to do). The implicit assumption is CDS | |
| 2518 | * ranges are assembled in order. Other cases should not use this method, | |
| 2519 | * but instead construct an explicit mapping for CDS (e.g. EMBL parsing). | |
| 2520 | */ | |
| 2521 | 8 | Collections.sort(result, IntRangeComparator.ASCENDING); |
| 2522 | 8 | return result; |
| 2523 | } | |
| 2524 | ||
| 2525 | /** | |
| 2526 | * Makes an alignment with a copy of the given sequences, adding in any | |
| 2527 | * non-redundant sequences which are mapped to by the cross-referenced | |
| 2528 | * sequences. | |
| 2529 | * | |
| 2530 | * @param seqs | |
| 2531 | * @param xrefs | |
| 2532 | * @param dataset | |
| 2533 | * the alignment dataset shared by the new copy | |
| 2534 | * @return | |
| 2535 | */ | |
| 2536 | 0 | public static AlignmentI makeCopyAlignment(SequenceI[] seqs, |
| 2537 | SequenceI[] xrefs, AlignmentI dataset) | |
| 2538 | { | |
| 2539 | 0 | AlignmentI copy = new Alignment(new Alignment(seqs)); |
| 2540 | 0 | copy.setDataset(dataset); |
| 2541 | 0 | boolean isProtein = !copy.isNucleotide(); |
| 2542 | 0 | SequenceIdMatcher matcher = new SequenceIdMatcher(seqs); |
| 2543 | 0 | if (xrefs != null) |
| 2544 | { | |
| 2545 | // BH 2019.01.25 recoded to remove iterators | |
| 2546 | ||
| 2547 | 0 | for (int ix = 0, nx = xrefs.length; ix < nx; ix++) |
| 2548 | { | |
| 2549 | 0 | SequenceI xref = xrefs[ix]; |
| 2550 | 0 | List<DBRefEntry> dbrefs = xref.getDBRefs(); |
| 2551 | 0 | if (dbrefs != null) |
| 2552 | { | |
| 2553 | 0 | for (int ir = 0, nir = dbrefs.size(); ir < nir; ir++) |
| 2554 | { | |
| 2555 | 0 | DBRefEntry dbref = dbrefs.get(ir); |
| 2556 | 0 | Mapping map = dbref.getMap(); |
| 2557 | 0 | SequenceI mto; |
| 2558 | 0 | if (map == null || (mto = map.getTo()) == null |
| 2559 | || mto.isProtein() != isProtein) | |
| 2560 | { | |
| 2561 | 0 | continue; |
| 2562 | } | |
| 2563 | 0 | SequenceI mappedTo = mto; |
| 2564 | 0 | SequenceI match = matcher.findIdMatch(mappedTo); |
| 2565 | 0 | if (match == null) |
| 2566 | { | |
| 2567 | 0 | matcher.add(mappedTo); |
| 2568 | 0 | copy.addSequence(mappedTo); |
| 2569 | } | |
| 2570 | } | |
| 2571 | } | |
| 2572 | } | |
| 2573 | } | |
| 2574 | 0 | return copy; |
| 2575 | } | |
| 2576 | ||
| 2577 | /** | |
| 2578 | * Try to align sequences in 'unaligned' to match the alignment of their | |
| 2579 | * mapped regions in 'aligned'. For example, could use this to align CDS | |
| 2580 | * sequences which are mapped to their parent cDNA sequences. | |
| 2581 | * | |
| 2582 | * This method handles 1:1 mappings (dna-to-dna or protein-to-protein). For | |
| 2583 | * dna-to-protein or protein-to-dna use alternative methods. | |
| 2584 | * | |
| 2585 | * @param unaligned | |
| 2586 | * sequences to be aligned | |
| 2587 | * @param aligned | |
| 2588 | * holds aligned sequences and their mappings | |
| 2589 | * @return | |
| 2590 | */ | |
| 2591 | 4 | public static int alignAs(AlignmentI unaligned, AlignmentI aligned) |
| 2592 | { | |
| 2593 | /* | |
| 2594 | * easy case - aligning a copy of aligned sequences | |
| 2595 | */ | |
| 2596 | 4 | if (alignAsSameSequences(unaligned, aligned)) |
| 2597 | { | |
| 2598 | 0 | return unaligned.getHeight(); |
| 2599 | } | |
| 2600 | ||
| 2601 | /* | |
| 2602 | * fancy case - aligning via mappings between sequences | |
| 2603 | */ | |
| 2604 | 4 | List<SequenceI> unmapped = new ArrayList<>(); |
| 2605 | 4 | Map<Integer, Map<SequenceI, Character>> columnMap = buildMappedColumnsMap( |
| 2606 | unaligned, aligned, unmapped); | |
| 2607 | 4 | int width = columnMap.size(); |
| 2608 | 4 | char gap = unaligned.getGapCharacter(); |
| 2609 | 4 | int realignedCount = 0; |
| 2610 | // TODO: verify this loop scales sensibly for very wide/high alignments | |
| 2611 | ||
| 2612 | 4 | for (SequenceI seq : unaligned.getSequences()) |
| 2613 | { | |
| 2614 | 26 | if (!unmapped.contains(seq)) |
| 2615 | { | |
| 2616 | 26 | char[] newSeq = new char[width]; |
| 2617 | 26 | Arrays.fill(newSeq, gap); // JBPComment - doubt this is faster than the |
| 2618 | // Integer iteration below | |
| 2619 | 26 | int newCol = 0; |
| 2620 | 26 | int lastCol = 0; |
| 2621 | ||
| 2622 | /* | |
| 2623 | * traverse the map to find columns populated | |
| 2624 | * by our sequence | |
| 2625 | */ | |
| 2626 | 26 | for (Integer column : columnMap.keySet()) |
| 2627 | { | |
| 2628 | 58976 | Character c = columnMap.get(column).get(seq); |
| 2629 | 58976 | if (c != null) |
| 2630 | { | |
| 2631 | /* | |
| 2632 | * sequence has a character at this position | |
| 2633 | * | |
| 2634 | */ | |
| 2635 | 31986 | newSeq[newCol] = c; |
| 2636 | 31986 | lastCol = newCol; |
| 2637 | } | |
| 2638 | 58976 | newCol++; |
| 2639 | } | |
| 2640 | ||
| 2641 | /* | |
| 2642 | * trim trailing gaps | |
| 2643 | */ | |
| 2644 | 26 | if (lastCol < width) |
| 2645 | { | |
| 2646 | 26 | char[] tmp = new char[lastCol + 1]; |
| 2647 | 26 | System.arraycopy(newSeq, 0, tmp, 0, lastCol + 1); |
| 2648 | 26 | newSeq = tmp; |
| 2649 | } | |
| 2650 | // TODO: optimise SequenceI to avoid char[]->String->char[] | |
| 2651 | 26 | seq.setSequence(String.valueOf(newSeq)); |
| 2652 | 26 | realignedCount++; |
| 2653 | } | |
| 2654 | } | |
| 2655 | 4 | return realignedCount; |
| 2656 | } | |
| 2657 | ||
| 2658 | /** | |
| 2659 | * If unaligned and aligned sequences share the same dataset sequences, then | |
| 2660 | * simply copies the aligned sequences to the unaligned sequences and returns | |
| 2661 | * true; else returns false | |
| 2662 | * | |
| 2663 | * @param unaligned | |
| 2664 | * - sequences to be aligned based on aligned | |
| 2665 | * @param aligned | |
| 2666 | * - 'guide' alignment containing sequences derived from same dataset | |
| 2667 | * as unaligned | |
| 2668 | * @return | |
| 2669 | */ | |
| 2670 | 8 | static boolean alignAsSameSequences(AlignmentI unaligned, |
| 2671 | AlignmentI aligned) | |
| 2672 | { | |
| 2673 | 8 | if (aligned.getDataset() == null || unaligned.getDataset() == null) |
| 2674 | { | |
| 2675 | 0 | return false; // should only pass alignments with datasets here |
| 2676 | } | |
| 2677 | ||
| 2678 | // map from dataset sequence to alignment sequence(s) | |
| 2679 | 8 | Map<SequenceI, List<SequenceI>> alignedDatasets = new HashMap<>(); |
| 2680 | 8 | for (SequenceI seq : aligned.getSequences()) |
| 2681 | { | |
| 2682 | 59 | SequenceI ds = seq.getDatasetSequence(); |
| 2683 | 59 | if (alignedDatasets.get(ds) == null) |
| 2684 | { | |
| 2685 | 58 | alignedDatasets.put(ds, new ArrayList<SequenceI>()); |
| 2686 | } | |
| 2687 | 59 | alignedDatasets.get(ds).add(seq); |
| 2688 | } | |
| 2689 | ||
| 2690 | /* | |
| 2691 | * first pass - check whether all sequences to be aligned share a | |
| 2692 | * dataset sequence with an aligned sequence; also note the leftmost | |
| 2693 | * ungapped column from which to copy | |
| 2694 | */ | |
| 2695 | 8 | int leftmost = Integer.MAX_VALUE; |
| 2696 | 8 | for (SequenceI seq : unaligned.getSequences()) |
| 2697 | { | |
| 2698 | 14 | final SequenceI ds = seq.getDatasetSequence(); |
| 2699 | 14 | if (!alignedDatasets.containsKey(ds)) |
| 2700 | { | |
| 2701 | 5 | return false; |
| 2702 | } | |
| 2703 | 9 | SequenceI alignedSeq = alignedDatasets.get(ds).get(0); |
| 2704 | 9 | int startCol = alignedSeq.findIndex(seq.getStart()); // 1.. |
| 2705 | 9 | leftmost = Math.min(leftmost, startCol); |
| 2706 | } | |
| 2707 | ||
| 2708 | /* | |
| 2709 | * second pass - copy aligned sequences; | |
| 2710 | * heuristic rule: pair off sequences in order for the case where | |
| 2711 | * more than one shares the same dataset sequence | |
| 2712 | */ | |
| 2713 | 3 | final char gapCharacter = aligned.getGapCharacter(); |
| 2714 | 3 | for (SequenceI seq : unaligned.getSequences()) |
| 2715 | { | |
| 2716 | 7 | List<SequenceI> alignedSequences = alignedDatasets |
| 2717 | .get(seq.getDatasetSequence()); | |
| 2718 | 7 | if (alignedSequences.isEmpty()) |
| 2719 | { | |
| 2720 | /* | |
| 2721 | * defensive check - shouldn't happen! (JAL-3536) | |
| 2722 | */ | |
| 2723 | 0 | continue; |
| 2724 | } | |
| 2725 | 7 | SequenceI alignedSeq = alignedSequences.get(0); |
| 2726 | ||
| 2727 | /* | |
| 2728 | * gap fill for leading (5') UTR if any | |
| 2729 | */ | |
| 2730 | // TODO this copies intron columns - wrong! | |
| 2731 | 7 | int startCol = alignedSeq.findIndex(seq.getStart()); // 1.. |
| 2732 | 7 | int endCol = alignedSeq.findIndex(seq.getEnd()); |
| 2733 | 7 | char[] seqchars = new char[endCol - leftmost + 1]; |
| 2734 | 7 | Arrays.fill(seqchars, gapCharacter); |
| 2735 | 7 | char[] toCopy = alignedSeq.getSequence(startCol - 1, endCol); |
| 2736 | 7 | System.arraycopy(toCopy, 0, seqchars, startCol - leftmost, |
| 2737 | toCopy.length); | |
| 2738 | 7 | seq.setSequence(String.valueOf(seqchars)); |
| 2739 | 7 | if (alignedSequences.size() > 0) |
| 2740 | { | |
| 2741 | // pop off aligned sequences (except the last one) | |
| 2742 | 7 | alignedSequences.remove(0); |
| 2743 | } | |
| 2744 | } | |
| 2745 | ||
| 2746 | /* | |
| 2747 | * finally remove gapped columns (e.g. introns) | |
| 2748 | */ | |
| 2749 | 3 | new RemoveGapColCommand("", unaligned.getSequencesArray(), 0, |
| 2750 | unaligned.getWidth() - 1, unaligned); | |
| 2751 | ||
| 2752 | 3 | return true; |
| 2753 | } | |
| 2754 | ||
| 2755 | /** | |
| 2756 | * Returns a map whose key is alignment column number (base 1), and whose | |
| 2757 | * values are a map of sequence characters in that column. | |
| 2758 | * | |
| 2759 | * @param unaligned | |
| 2760 | * @param aligned | |
| 2761 | * @param unmapped | |
| 2762 | * @return | |
| 2763 | */ | |
| 2764 | 4 | static SortedMap<Integer, Map<SequenceI, Character>> buildMappedColumnsMap( |
| 2765 | AlignmentI unaligned, AlignmentI aligned, | |
| 2766 | List<SequenceI> unmapped) | |
| 2767 | { | |
| 2768 | /* | |
| 2769 | * Map will hold, for each aligned column position, a map of | |
| 2770 | * {unalignedSequence, characterPerSequence} at that position. | |
| 2771 | * TreeMap keeps the entries in ascending column order. | |
| 2772 | */ | |
| 2773 | 4 | SortedMap<Integer, Map<SequenceI, Character>> map = new TreeMap<>(); |
| 2774 | ||
| 2775 | /* | |
| 2776 | * record any sequences that have no mapping so can't be realigned | |
| 2777 | */ | |
| 2778 | 4 | unmapped.addAll(unaligned.getSequences()); |
| 2779 | ||
| 2780 | 4 | List<AlignedCodonFrame> mappings = aligned.getCodonFrames(); |
| 2781 | ||
| 2782 | 4 | for (SequenceI seq : unaligned.getSequences()) |
| 2783 | { | |
| 2784 | 26 | for (AlignedCodonFrame mapping : mappings) |
| 2785 | { | |
| 2786 | 510 | SequenceI fromSeq = mapping.findAlignedSequence(seq, aligned); |
| 2787 | 510 | if (fromSeq != null) |
| 2788 | { | |
| 2789 | 26 | Mapping seqMap = mapping.getMappingBetween(fromSeq, seq); |
| 2790 | 26 | if (addMappedPositions(seq, fromSeq, seqMap, map)) |
| 2791 | { | |
| 2792 | 26 | unmapped.remove(seq); |
| 2793 | } | |
| 2794 | } | |
| 2795 | } | |
| 2796 | } | |
| 2797 | 4 | return map; |
| 2798 | } | |
| 2799 | ||
| 2800 | /** | |
| 2801 | * Helper method that adds to a map the mapped column positions of a sequence. | |
| 2802 | * <br> | |
| 2803 | * For example if aaTT-Tg-gAAA is mapped to TTTAAA then the map should record | |
| 2804 | * that columns 3,4,6,10,11,12 map to characters T,T,T,A,A,A of the mapped to | |
| 2805 | * sequence. | |
| 2806 | * | |
| 2807 | * @param seq | |
| 2808 | * the sequence whose column positions we are recording | |
| 2809 | * @param fromSeq | |
| 2810 | * a sequence that is mapped to the first sequence | |
| 2811 | * @param seqMap | |
| 2812 | * the mapping from 'fromSeq' to 'seq' | |
| 2813 | * @param map | |
| 2814 | * a map to add the column positions (in fromSeq) of the mapped | |
| 2815 | * positions of seq | |
| 2816 | * @return | |
| 2817 | */ | |
| 2818 | 28 | static boolean addMappedPositions(SequenceI seq, SequenceI fromSeq, |
| 2819 | Mapping seqMap, Map<Integer, Map<SequenceI, Character>> map) | |
| 2820 | { | |
| 2821 | 28 | if (seqMap == null) |
| 2822 | { | |
| 2823 | 0 | return false; |
| 2824 | } | |
| 2825 | ||
| 2826 | /* | |
| 2827 | * invert mapping if it is from unaligned to aligned sequence | |
| 2828 | */ | |
| 2829 | 28 | if (seqMap.getTo() == fromSeq.getDatasetSequence()) |
| 2830 | { | |
| 2831 | 0 | seqMap = new Mapping(seq.getDatasetSequence(), |
| 2832 | seqMap.getMap().getInverse()); | |
| 2833 | } | |
| 2834 | ||
| 2835 | 28 | int toStart = seq.getStart(); |
| 2836 | ||
| 2837 | /* | |
| 2838 | * traverse [start, end, start, end...] ranges in fromSeq | |
| 2839 | */ | |
| 2840 | 28 | for (int[] fromRange : seqMap.getMap().getFromRanges()) |
| 2841 | { | |
| 2842 | 62 | for (int i = 0; i < fromRange.length - 1; i += 2) |
| 2843 | { | |
| 2844 | 31 | boolean forward = fromRange[i + 1] >= fromRange[i]; |
| 2845 | ||
| 2846 | /* | |
| 2847 | * find the range mapped to (sequence positions base 1) | |
| 2848 | */ | |
| 2849 | 31 | int[] range = seqMap.locateMappedRange(fromRange[i], |
| 2850 | fromRange[i + 1]); | |
| 2851 | 31 | if (range == null) |
| 2852 | { | |
| 2853 | 0 | jalview.bin.Console.errPrintln("Error in mapping " + seqMap |
| 2854 | + " from " + fromSeq.getName()); | |
| 2855 | 0 | return false; |
| 2856 | } | |
| 2857 | 31 | int fromCol = fromSeq.findIndex(fromRange[i]); |
| 2858 | 31 | int mappedCharPos = range[0]; |
| 2859 | ||
| 2860 | /* | |
| 2861 | * walk over the 'from' aligned sequence in forward or reverse | |
| 2862 | * direction; when a non-gap is found, record the column position | |
| 2863 | * of the next character of the mapped-to sequence; stop when all | |
| 2864 | * the characters of the range have been counted | |
| 2865 | */ | |
| 2866 | 2794274 | while (mappedCharPos <= range[1] && fromCol <= fromSeq.getLength() |
| 2867 | && fromCol >= 0) | |
| 2868 | { | |
| 2869 | 2794243 | if (!Comparison.isGap(fromSeq.getCharAt(fromCol - 1))) |
| 2870 | { | |
| 2871 | /* | |
| 2872 | * mapped from sequence has a character in this column | |
| 2873 | * record the column position for the mapped to character | |
| 2874 | */ | |
| 2875 | 31998 | Map<SequenceI, Character> seqsMap = map.get(fromCol); |
| 2876 | 31998 | if (seqsMap == null) |
| 2877 | { | |
| 2878 | 5398 | seqsMap = new HashMap<>(); |
| 2879 | 5398 | map.put(fromCol, seqsMap); |
| 2880 | } | |
| 2881 | 31998 | seqsMap.put(seq, seq.getCharAt(mappedCharPos - toStart)); |
| 2882 | 31998 | mappedCharPos++; |
| 2883 | } | |
| 2884 | 2794243 | fromCol += (forward ? 1 : -1); |
| 2885 | } | |
| 2886 | } | |
| 2887 | } | |
| 2888 | 28 | return true; |
| 2889 | } | |
| 2890 | ||
| 2891 | // strictly temporary hack until proper criteria for aligning protein to cds | |
| 2892 | // are in place; this is so Ensembl -> fetch xrefs Uniprot aligns the Uniprot | |
| 2893 | 4 | public static boolean looksLikeEnsembl(AlignmentI alignment) |
| 2894 | { | |
| 2895 | 4 | for (SequenceI seq : alignment.getSequences()) |
| 2896 | { | |
| 2897 | 88 | String name = seq.getName(); |
| 2898 | 88 | if (!name.startsWith("ENSG") && !name.startsWith("ENST")) |
| 2899 | { | |
| 2900 | 0 | return false; |
| 2901 | } | |
| 2902 | } | |
| 2903 | 4 | return true; |
| 2904 | } | |
| 2905 | ||
| 2906 | 5 | public static boolean isSecondaryStructurePresent( |
| 2907 | AlignmentAnnotation[] annotations) | |
| 2908 | { | |
| 2909 | 5 | boolean ssPresent = false; |
| 2910 | ||
| 2911 | 5 | for (AlignmentAnnotation aa : annotations) |
| 2912 | { | |
| 2913 | 5 | if (ssPresent) |
| 2914 | { | |
| 2915 | 0 | break; |
| 2916 | } | |
| 2917 | ||
| 2918 | 5 | if (Constants.SECONDARY_STRUCTURE_LABELS.containsKey(aa.label)) |
| 2919 | { | |
| 2920 | 3 | ssPresent = true; |
| 2921 | 3 | break; |
| 2922 | } | |
| 2923 | } | |
| 2924 | ||
| 2925 | 5 | return ssPresent; |
| 2926 | ||
| 2927 | } | |
| 2928 | ||
| 2929 | 0 | public static Color getSecondaryStructureAnnotationColour(char symbol) |
| 2930 | { | |
| 2931 | ||
| 2932 | 0 | if (symbol == Constants.COIL) |
| 2933 | { | |
| 2934 | 0 | return Color.gray; |
| 2935 | } | |
| 2936 | 0 | if (symbol == Constants.SHEET) |
| 2937 | { | |
| 2938 | 0 | return Color.green; |
| 2939 | } | |
| 2940 | 0 | if (symbol == Constants.HELIX) |
| 2941 | { | |
| 2942 | 0 | return Color.red; |
| 2943 | } | |
| 2944 | ||
| 2945 | 0 | return Color.white; |
| 2946 | } | |
| 2947 | ||
| 2948 | 70935 | public static char findSSAnnotationForGivenSeqposition( |
| 2949 | AlignmentAnnotation aa, int seqPosition) | |
| 2950 | { | |
| 2951 | 70935 | char ss = '*'; |
| 2952 | ||
| 2953 | 70936 | if (aa != null) |
| 2954 | { | |
| 2955 | 70935 | if (aa.getAnnotationForPosition(seqPosition) != null) |
| 2956 | { | |
| 2957 | 41675 | Annotation a = aa.getAnnotationForPosition(seqPosition); |
| 2958 | 41674 | ss = a.secondaryStructure; |
| 2959 | ||
| 2960 | // There is no representation for coil and it can be either ' ' or null. | |
| 2961 | 41676 | if (ss == ' ' || ss == '-') |
| 2962 | { | |
| 2963 | 9746 | ss = Constants.COIL; |
| 2964 | } | |
| 2965 | } | |
| 2966 | else | |
| 2967 | { | |
| 2968 | 29260 | ss = Constants.COIL; |
| 2969 | } | |
| 2970 | } | |
| 2971 | ||
| 2972 | 70936 | return ss; |
| 2973 | } | |
| 2974 | ||
| 2975 | 1691 | public static List<String> extractSSSourceInAlignmentAnnotation( |
| 2976 | AlignmentAnnotation[] annotations) | |
| 2977 | { | |
| 2978 | ||
| 2979 | 1691 | List<String> ssSources = new ArrayList<>(); |
| 2980 | 1691 | Set<String> addedSources = new HashSet<>(); // to keep track of added |
| 2981 | // sources | |
| 2982 | ||
| 2983 | 1691 | if (annotations == null) |
| 2984 | { | |
| 2985 | 12 | return ssSources; |
| 2986 | } | |
| 2987 | ||
| 2988 | 1679 | for (AlignmentAnnotation aa : annotations) |
| 2989 | { | |
| 2990 | ||
| 2991 | 7873 | String ssSource = extractSSSourceFromAnnotationDescription(aa); |
| 2992 | ||
| 2993 | 7873 | if (ssSource != null && !addedSources.contains(ssSource)) |
| 2994 | { | |
| 2995 | 68 | ssSources.add(ssSource); |
| 2996 | 68 | addedSources.add(ssSource); |
| 2997 | } | |
| 2998 | ||
| 2999 | } | |
| 3000 | 1679 | Collections.sort(ssSources); |
| 3001 | ||
| 3002 | 1679 | return ssSources; |
| 3003 | ||
| 3004 | } | |
| 3005 | ||
| 3006 | 62816 | public static String extractSSSourceFromAnnotationDescription( |
| 3007 | AlignmentAnnotation aa) | |
| 3008 | { | |
| 3009 | ||
| 3010 | 62816 | for (String label : Constants.SECONDARY_STRUCTURE_LABELS.keySet()) |
| 3011 | { | |
| 3012 | ||
| 3013 | 70055 | if (label.equals(aa.label)) |
| 3014 | { | |
| 3015 | ||
| 3016 | 55577 | if (aa.getProperty(Constants.SS_PROVIDER_PROPERTY) != null) |
| 3017 | { | |
| 3018 | ||
| 3019 | 0 | return aa.getProperty(Constants.SS_PROVIDER_PROPERTY); |
| 3020 | ||
| 3021 | } | |
| 3022 | ||
| 3023 | // For JPred | |
| 3024 | 55576 | if (Constants.SS_ANNOTATION_FROM_JPRED_LABEL.equals(aa.label)) |
| 3025 | { | |
| 3026 | ||
| 3027 | 0 | return (Constants.SECONDARY_STRUCTURE_LABELS.get(aa.label)); |
| 3028 | ||
| 3029 | } | |
| 3030 | ||
| 3031 | // For input with secondary structure | |
| 3032 | 55576 | if (Constants.SS_ANNOTATION_LABEL.equals(aa.label) |
| 3033 | && aa.description != null | |
| 3034 | && Constants.SS_ANNOTATION_LABEL.equals(aa.description)) | |
| 3035 | { | |
| 3036 | ||
| 3037 | 17328 | return (Constants.SECONDARY_STRUCTURE_LABELS.get(aa.label)); |
| 3038 | ||
| 3039 | } | |
| 3040 | ||
| 3041 | // For other sources | |
| 3042 | 38248 | if (aa.sequenceRef == null) |
| 3043 | { | |
| 3044 | 174 | return null; |
| 3045 | } | |
| 3046 | 38074 | else if (aa.sequenceRef.getDatasetSequence() == null) |
| 3047 | { | |
| 3048 | 0 | return null; |
| 3049 | } | |
| 3050 | 38074 | Vector<PDBEntry> pdbEntries = aa.sequenceRef.getDatasetSequence() |
| 3051 | .getAllPDBEntries(); | |
| 3052 | ||
| 3053 | // TODO: this is an incredibly fragile mechanism | |
| 3054 | 38074 | for (PDBEntry entry : pdbEntries) |
| 3055 | { | |
| 3056 | ||
| 3057 | 46188 | String entryProvider = entry.getProvider(); |
| 3058 | 46188 | if (entryProvider == null) |
| 3059 | { | |
| 3060 | // No provider - so this is either an old Jalview project, or not | |
| 3061 | // retrieved from recognised source | |
| 3062 | 46188 | entryProvider = "PDB"; |
| 3063 | } | |
| 3064 | ||
| 3065 | // Should (re)use a standard mechanism for extracting the PDB ID as it | |
| 3066 | // is written 1QWXTUV:CHAIN | |
| 3067 | // Trim the string from first occurrence of colon | |
| 3068 | 46188 | String entryID = entry.getId(); |
| 3069 | 46188 | int index = entryID.indexOf(':'); |
| 3070 | ||
| 3071 | // Check if colon exists | |
| 3072 | 46188 | if (index != -1) |
| 3073 | { | |
| 3074 | ||
| 3075 | // Trim the string from first occurrence of colon | |
| 3076 | 0 | entryID = entryID.substring(0, index); |
| 3077 | ||
| 3078 | } | |
| 3079 | ||
| 3080 | // TODO: shouldn't need to extract from description what the | |
| 3081 | // originating ID is for this annotation! | |
| 3082 | 46188 | if (entryProvider == "PDB" && aa.description.toLowerCase() |
| 3083 | .contains("secondary structure for " | |
| 3084 | + entryID.toLowerCase())) | |
| 3085 | { | |
| 3086 | ||
| 3087 | 38074 | return entryProvider; |
| 3088 | ||
| 3089 | } | |
| 3090 | ||
| 3091 | 8114 | else if (entryProvider != "PDB" && aa.description.toLowerCase() |
| 3092 | .contains(entryID.toLowerCase())) | |
| 3093 | { | |
| 3094 | ||
| 3095 | 0 | return entryProvider; |
| 3096 | ||
| 3097 | } | |
| 3098 | ||
| 3099 | } | |
| 3100 | } | |
| 3101 | } | |
| 3102 | ||
| 3103 | 7239 | return null; |
| 3104 | ||
| 3105 | } | |
| 3106 | ||
| 3107 | // to do set priority for labels | |
| 3108 | 10821642 | public static List<AlignmentAnnotation> getAlignmentAnnotationForSource( |
| 3109 | SequenceI seq, String ssSource) | |
| 3110 | { | |
| 3111 | ||
| 3112 | 10822917 | List<AlignmentAnnotation> ssAnnots = new ArrayList<AlignmentAnnotation>(); |
| 3113 | 10882691 | for (String ssLabel : Constants.SECONDARY_STRUCTURE_LABELS.keySet()) |
| 3114 | { | |
| 3115 | ||
| 3116 | 21706654 | AlignmentAnnotation[] aa = seq.getAnnotation(ssLabel); |
| 3117 | 21441485 | if (aa != null) |
| 3118 | { | |
| 3119 | ||
| 3120 | 101130 | if (Constants.SS_ALL_PROVIDERS.equals(ssSource)) |
| 3121 | { | |
| 3122 | 50571 | ssAnnots.addAll(Arrays.asList(aa)); |
| 3123 | 50571 | continue; |
| 3124 | } | |
| 3125 | ||
| 3126 | 50559 | for (AlignmentAnnotation annot : aa) |
| 3127 | { | |
| 3128 | ||
| 3129 | 54943 | String ssSourceForAnnot = extractSSSourceFromAnnotationDescription( |
| 3130 | annot); | |
| 3131 | 54943 | if (ssSourceForAnnot != null && ssSource.equals(ssSourceForAnnot)) |
| 3132 | { | |
| 3133 | 54943 | ssAnnots.add(annot); |
| 3134 | } | |
| 3135 | } | |
| 3136 | } | |
| 3137 | } | |
| 3138 | 10761489 | if (ssAnnots.size() > 0) |
| 3139 | { | |
| 3140 | 101127 | return ssAnnots; |
| 3141 | } | |
| 3142 | ||
| 3143 | 10686191 | return null; |
| 3144 | ||
| 3145 | } | |
| 3146 | ||
| 3147 | 3 | public static Map<SequenceI, ArrayList<AlignmentAnnotation>> getSequenceAssociatedAlignmentAnnotations( |
| 3148 | AlignmentAnnotation[] alignAnnotList, String selectedSSSource) | |
| 3149 | { | |
| 3150 | ||
| 3151 | 3 | Map<SequenceI, ArrayList<AlignmentAnnotation>> ssAlignmentAnnotationForSequences = new HashMap<SequenceI, ArrayList<AlignmentAnnotation>>(); |
| 3152 | 3 | if (alignAnnotList == null || alignAnnotList.length == 0) |
| 3153 | { | |
| 3154 | 0 | return ssAlignmentAnnotationForSequences; |
| 3155 | } | |
| 3156 | ||
| 3157 | 3 | for (AlignmentAnnotation aa : alignAnnotList) |
| 3158 | { | |
| 3159 | 12 | if (aa.sequenceRef == null) |
| 3160 | { | |
| 3161 | 12 | continue; |
| 3162 | } | |
| 3163 | ||
| 3164 | 0 | if (isSecondaryStructureFrom(selectedSSSource, aa)) |
| 3165 | { | |
| 3166 | 0 | ssAlignmentAnnotationForSequences |
| 3167 | .computeIfAbsent(aa.sequenceRef.getDatasetSequence(), | |
| 3168 | k -> new ArrayList<>()) | |
| 3169 | .add(aa); | |
| 3170 | } | |
| 3171 | } | |
| 3172 | ||
| 3173 | 3 | return ssAlignmentAnnotationForSequences; |
| 3174 | ||
| 3175 | } | |
| 3176 | ||
| 3177 | /** | |
| 3178 | * | |
| 3179 | * @param selectedSSSource | |
| 3180 | * @param aa | |
| 3181 | * @return true if aa is from a provider or all providers as specified by | |
| 3182 | * selectedSSSource | |
| 3183 | */ | |
| 3184 | 0 | public static boolean isSecondaryStructureFrom(String selectedSSSource, |
| 3185 | AlignmentAnnotation aa) | |
| 3186 | { | |
| 3187 | ||
| 3188 | 0 | for (String label : Constants.SECONDARY_STRUCTURE_LABELS.keySet()) |
| 3189 | { | |
| 3190 | ||
| 3191 | 0 | if (label.equals(aa.label)) |
| 3192 | { | |
| 3193 | ||
| 3194 | 0 | if (selectedSSSource.equals(Constants.SS_ALL_PROVIDERS)) |
| 3195 | { | |
| 3196 | 0 | return true; |
| 3197 | } | |
| 3198 | 0 | String ssSource = AlignmentUtils |
| 3199 | .extractSSSourceFromAnnotationDescription(aa); | |
| 3200 | 0 | if (ssSource != null && ssSource.equals(selectedSSSource)) |
| 3201 | { | |
| 3202 | 0 | return true; |
| 3203 | } | |
| 3204 | } | |
| 3205 | } | |
| 3206 | 0 | return false; |
| 3207 | } | |
| 3208 | ||
| 3209 | // Method to get the key for a given provider value | |
| 3210 | 0 | public static String getSecondaryStructureProviderKey(String providerValue) { |
| 3211 | 0 | for (Map.Entry<String, String> entry : Constants.STRUCTURE_PROVIDERS.entrySet()) { |
| 3212 | 0 | if (entry.getValue().equals(providerValue)) { |
| 3213 | 0 | return entry.getKey(); // Return the key (abbreviation) for the matching provider value |
| 3214 | } | |
| 3215 | } | |
| 3216 | 0 | return null; // Return null if no match is found |
| 3217 | } | |
| 3218 | ||
| 3219 | 0 | public static String reduceLabelLength(String label) { |
| 3220 | // Split the input by " | " | |
| 3221 | 0 | String[] parts = label.split(" \\| "); |
| 3222 | ||
| 3223 | // Map the full names to their abbreviations | |
| 3224 | 0 | String reducedLabel = Arrays.stream(parts) |
| 3225 | .map(fullName -> Constants.STRUCTURE_PROVIDERS.entrySet().stream() | |
| 3226 | .filter(entry -> entry.getValue().equals(fullName)) | |
| 3227 | .map(Map.Entry::getKey) | |
| 3228 | .findFirst() | |
| 3229 | .orElse(fullName)) // Use fullName if no abbreviation is found | |
| 3230 | .collect(Collectors.joining(" | ")); | |
| 3231 | ||
| 3232 | 0 | return reducedLabel; // Return the reduced label if abbreviations were applied |
| 3233 | } | |
| 3234 | ||
| 3235 | 0 | public static Color getSecondaryStructureProviderColor(String label) { |
| 3236 | ||
| 3237 | //return Constants.STRUCTURE_PROVIDERS_COLOR.getOrDefault(label, Color.BLACK); | |
| 3238 | 0 | Color c = Constants.STRUCTURE_PROVIDERS_COLOR.get(label.trim()); |
| 3239 | 0 | if(c==null) |
| 3240 | 0 | c = Color.BLACK; |
| 3241 | 0 | return c; |
| 3242 | } | |
| 3243 | ||
| 3244 | ||
| 3245 | 0 | public static void assignSecondaryStructureProviderColor(Map<String, Color> secondaryStructureProviderColorMap, |
| 3246 | List<String> labels) { | |
| 3247 | ||
| 3248 | // Use a Set to track unique labels | |
| 3249 | 0 | Set<String> uniqueLabels = new HashSet<>(labels); |
| 3250 | ||
| 3251 | 0 | Color[] palette = ColorBrewer.Paired.getColorPalette(uniqueLabels.size()); |
| 3252 | ||
| 3253 | ||
| 3254 | 0 | List<Color> colorList = new ArrayList<>(); |
| 3255 | 0 | Collections.addAll(colorList, palette); |
| 3256 | 0 | Collections.shuffle(colorList); |
| 3257 | 0 | int i = 0; |
| 3258 | ||
| 3259 | // Loop through each unique label and add it to the map with a color. | |
| 3260 | 0 | for (String label : uniqueLabels) { |
| 3261 | // Generate or retrieve a color for the label. | |
| 3262 | 0 | secondaryStructureProviderColorMap.put(label.toUpperCase().trim(), colorList.get(i)); |
| 3263 | 0 | i++; |
| 3264 | } | |
| 3265 | } | |
| 3266 | } |